










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.31 no.1 Bogotá Jan./Mar. 2016
Recurrent Benign Intrahepatic Cholestasis is a Diagnostic Challenge
Pedro Sánchez Márquez MD. (1), Mario H. Rey Tovar MD. (2), Martín A. Garzón MD. (2), Tatiana Echeverry MD. (3)
(1) Internal Medicine Resident at the Universidad de la Sabana in Bogotá, Colombia.
(2) Internist and Gastroenterologist at the Hospital Universitario la Samaritana in Bogotá, Colombia.
(3) Internist at the Hospital Universitario la Samaritana in Bogotá, Colombia.
Received: 23-09-15 Accepted: 26-01-16
Abstract
Hepatic cholestasis includes a large variety of disorders which can compromise the intrahepatic and extrahepatic pathways. Diagnosis requires a combination of clinical, biochemical, imaging, and sometimes pathological, findings. We describe the case of a patient with intermittent episodes of jaundice which resolved by themselves but which were and decisive associated with abdominal pain and severe itching. These episodes occurred during exacerbation of the intrahepatic cholestatic pattern but completely resolved during episodes of remission.
Keywords
Cholestasis, benign recurrent intrahepatic cholestasis, progressive familial intrahepatic cholestasis, jaundice, pruritus.
INTRODUCTION
Jaundice is a common presentation of a variety of hepatobiliary disorders which results from overproduction of bilirubin, changes in conjugation, biliary obstruction and liver inflammation. (1)
Cholestasis is a clinical and biochemical syndrome caused by an alteration in the flow of bile resulting from biliary tract obstruction or an alteration in uptake, conjugation or biliary acid extraction. (2) It can be intrahepatic when compromise of biliary excretion occurs within the hepatocellular cytoplasm and in medium sized bile ducts (up to 400 microns in diameter), or it can be extrahepatic when major bile ducts, including the hepatic bile duct and common bile duct are compromised. (3)
Cholestasis is diagnosed on the basis of clinical findings and confirmed when alterations in liver biochemistry is found, when images show bile duct obstruction, or in some bases by examination of liver biopsies.
Independent of its cause, cholestasis most frequently manifests as jaundice and itching which affect the patient quality of life.
CASE REPORT
The patient in this case was a 55 year old single man who worked as a farm laborer. Every six months he had developed episodes of generalized jaundice, nausea, itching, dark urine, and biting right upper quadrant abdominal pain. The episodes developed slowly and progressively and then receded until resolution in the same manner without triggering or mitigating events. Symptoms completely disappeared between episodes. The patient had a personal history of ingesting alcohol more than 30 grams of alcohol per week for 30 years, previous hospitalizations for jaundice, a hearing disability from childhood, and had had an open cholecystectomy because of gall stones in 2003.
Physical examination showed generally acceptable patient condition with generalized mucocutaneous jaundice but without no signs of chronic liver disease or portal hypertension.
During previous episodes of alternating jaundice and remission, the patient had undergone many diagnostic studies including endoscopic retrograde cholangiopancreatography and total abdominal ultrasound. All reported surgical absence of gallbladder without intrahepatic or extrahepatic bile duct alterations. Paraclinical tests showed a cholestatic pattern of jaundice during the acute phase and a normal pattern during remission. A liver biopsy taken in 2003 showed intact architecture, pericentral canalicular cholestasis with histiocytes, but no inflammation within the bile ducts.
The patients most recent laboratory test results during the acute phase are: CBC: Normal; INR: 1.11; Albumin: 2.41 mg/dL; Transferrin: Normal; Ceruloplasmin: Normal; TIBC: Normal; TSH: Normal; CA 19-9: Negative; Kidney function: Normal; AMA: Negative; ANA: Negative; ASMA: Positive 1:80 dilutions; Total Bilirubin: 30.1 mg/dL; Indirect Bilirubin; Direct Bilirubin: 19.3 mg/dL; AST: AST 41.3 U/L; ALT: 31.2 U/L; anti-LKM: Negative; ENAS: Negative; Alkaline phosphatase: 407 IU/L; Alpha-fetoprotein: Negative; Hepatitis B: Nonreactive; Hepatitis C: Nonreactive; Prothrombin Time: 13.7/12.3; PTT: 27.1/25.2;Serum iron: Normal.
An abdominal CT showed which showed a fatty liver, no evidence of focal lesions, normal intrahepatic and extrahepatic bile ducts, and no gallbladder. The spleen, pancreas, adrenal glands, kidneys and collection systems all appeared to be normal. The retroperitoneum showed no lesions or disorders (Figures 1).
A new liver biopsy showed hepatic architecture preserved, with severe pericentral intrahepatic cholestasis, degeneration of vacuoles in hepatocytes with biliary pigment, no evidence of injury to the portal spaces (Figures 2, 3, and 4).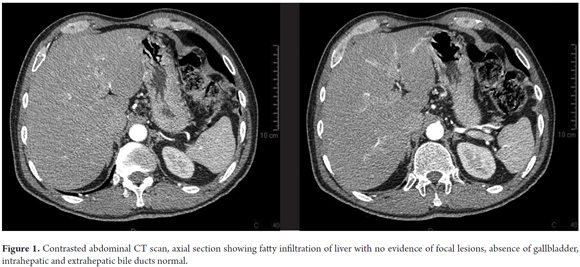
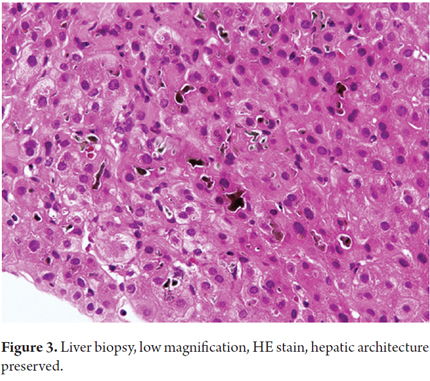
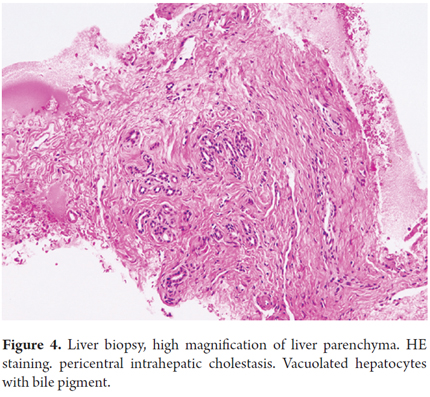
It was concluded that, in the absence of cirrhosis, biopsy and laboratory results together with recurrent episodes of jaundice indicated benign intrahepatic cholestasis.
LITERATURE REVIEW
Benign recurrent intrahepatic cholestasis (BRIC) is a rare type of cholestasis. It was first described in 1959 by Summerskill and Walshe. (2)
It is more common in men than in women and usually develops after the first decade of life. It sometimes recurs at intervals of anywhere from one year to a decade with episodes that that last from weeks to months and then resolve spontaneously without leading to progressive hepatocellular dysfunction, fibrosis or cirrhosis. (4, 5)
The diagnostic criteria established by Luketic and Shiffman include (6):
1. A history of at least two episodes of jaundice with asymptomatic periods of months to years in between.
2. Paraclinical test results consistent with intrahepatic cholestasis, including elevated alkaline phosphatase, conjugated hyperbilirubinemia, GGT and aminotransferase at the upper limit.
3. Intense itching secondary to cholestasis, although this may be absent in 25% of cases.
4. No other known etiological factors for cholestasis.
5. Normal intra- and extrahepatic biliary duct morphology demonstrated by ERCP.
6. Liver biopsy showing centrilobular cholestasis.
All these resolve in between acute periods. It has an autosomal recessive inheritance pattern, with incomplete penetrance. It is associated with mutations in genes ATP8B1 (I) and ABCB11 (II) located on chromosome 18 (18q21-22) and 2 respectively that are involved in progressive familial intrahepatic cholestasis. (7)
In patients with BRIC, these mutations are associated with alterations in the ATPase involved in the transport of cations such as copper, calcium, sodium and potassium and in the positioning of the phospholipids that make up the cell membranes. These modifications are associated with changes in the permeability of the membrane which lead to disturbances in cell transport and consequent reductions in the secretion of bile acids in the liver, pancreatic exocrine secretions and reabsorption of bile salts in the intestine. Taken together, they all lead to increasing losses of salts characteristic of BRIC patients. (8, 9)
As of today there are no specific treatments available for prevention or reduction of the duration of attacks. Primary treatment is based on relieving symptoms until each episode spontaneously resolves. Cholestyramine and ursodeoxycholic acid have been used because they protect cell membranes, prevent cytolysis and apoptosis induced by bile acids, and have an immunomodulatory effect that decreases the release of IL-2, IL-4 and interferon. (10)
When cirrhosis does not develop, the prognosis is good. As an attack resolves itself, pruritus also resolves quickly and completely as do jaundice and hepatic enzyme abnormalities. (4, 5)
REFERENCES
1. James W, Aaron M. Diagnostic Approach to the Patient with Jaundice. Prim Care Clin Office Pract. 2011;38:469-482. [ Links ]
2. Ken D, Vinay S, Walid S. Atypical causes of cholestasis. World J Gastroentero. 2014; July 28; 20 (28): 9418-9426. [ Links ]
3. Pérez T, López P, Tomás E., et al. Diagnostic and therapeutic approach to cholestatic liver disease. Rev Espa Enferm Dig. 2004;96:1:60-73. [ Links ]
4. Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Cli Liver Dis 2004;8:133-49. [ Links ]
5. Van der Woerd WL, van Mil SW, et al. Familial cholestasis: Progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis. Best Pract Res Clin Gastroenterol. 2010;24:541. [ Links ]
6. Renard R, Geubel AP, Benhamou JP. Bening recurrent intrahepatic cholestasis. J Clin Gastroenterol. 1989;11:546-51. [ Links ]
7. Anshu Srivastava. Progressive Familial Intrahepatic Cholestasis. J Clin Exper Hepatol. 2014;3:4:25-36. [ Links ]
8. Minuk GY, Shaffer EA. Bening recurrent intrahepatic cholestasis. Evidence for an intrinsic abnormality in hepatocite secretion. Gastroenterology. 1987;93:1187-93. [ Links ]
9. Bijleveld CM, Vonk RJ, Kuipers F, Havinga R, et al. Bening recurrent intrahepatica cholestasis: Altered bile acid metabolism. Gastroenterology. 1989;97:427-32. [ Links ]
10. Maggiore G. Efficacy of ursodeoxycholic acid in preventing cholestasic episodes in a patient with bening recurrent intrahepatic cholestasis. Hepatology. 1992;16:504. [ Links ]
