











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El hígado es un órgano vital que realiza diversos procesos metabólicos que van desde la transformación y eliminación de toxinas, hasta el almacenamiento y producción de biomoléculas indispensables para la vida. Se denomina insuficiencia o falla hepática cuando el hígado está incapacitado para llevar a cabo sus funciones de síntesis y metabolismo; inicialmente se puede clasificar la falla hepática como aguda o crónica, y recientemente se reconoce un nuevo estado caracterizado por una exacerbación aguda sobre un daño hepático crónico 1,2.
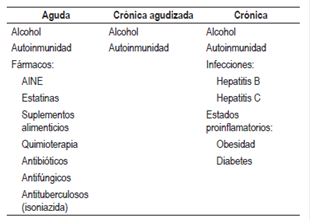
La lesión inducida por fármacos es la principal causa de falla hepática, que se subdivide de acuerdo con el tipo de daño ocasionado: intrínseca (dependiente de la dosis), cuyo mejor ejemplo es el acetaminofén; e idiosincrática (independiente de la dosis) 3,4,5. Las causas más frecuentes de falla hepática crónica son la enfermedad hepática por alcohol, seguida de la ocasionada por agentes infecciosos (75%-80% asociada con el virus de la hepatitis C -VHC-). Algunos expertos consideran que los estados proinflamatorios asociados con esteatosis, como la obesidad o la diabetes, también deben ser considerados como factores para el desarrollo de falla hepática crónica. Las enfermedades autoinmunes merecen mención particular porque pueden manifestarse con una insuficiencia hepática aguda, o causar un deterioro crónico (Tabla 1) 6,7.
Fisiología de la hemostasia
La hemostasia es un proceso complejo que involucra la estimulación de factores de coagulación sobre una superficie de plaquetas activadas para lograr la reparación del tejido vascular 8. En general, la hemostasia se divide en primaria, mediada por el endotelio y las plaquetas con el fin de integrar el tapón hemostático; y secundaria, que implica el proceso de coagulación 8,9.
Durante las últimas décadas se ha logrado tener una mejor comprensión de la hemostasia. Uno de los cambios más relevantes es dejar de considerar a las plaquetas únicamente como los elementos formes que integran el tapón plaquetario durante la hemostasia primaria, ahora se sabe que poseen más de 300 sustancias contenidas en los gránulos intracelulares (gránulos α), entre los que se destacan los factores de crecimiento necesarios para la regeneración tisular 10.
Modelo celular de la coagulación
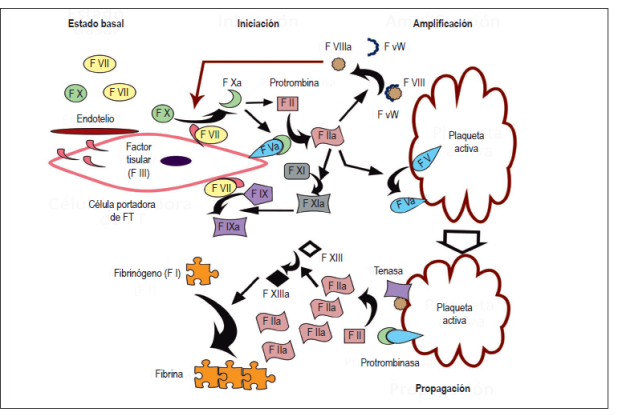
Tradicionalmente, la coagulación era entendida como una activación ordenada de factores en forma de cascada, pero actualmente el modelo propuesto por Hoffman y Monroe permite una mejor comprensión al considerar las interacciones entre los factores y los elementos celulares 11. El modelo celular de la coagulación parte del estado basal donde el factor X y VII se encuentran inactivos en el torrente sanguíneo y existen células portadoras (endotelio, subendotelio y monocitos) del factor tisular (FT, o también conocido como factor III), ante los estímulos nocivos el FT es liberado al torrente sanguíneo y comienza propiamente el proceso de la coagulación que consta de 3 fases:
Fase 1 o iniciación: el FT se une al factor VII para transformar y activar los factores IX y X. El factor Xa se combina con el factor Va presente en las superficies celulares, transformando pequeñas cantidades de protrombina (factor II) a trombina (factor IIa).
Fase 2 o amplificación: la trombina mantiene la producción del factor Xa mediante su intervención en la vía intrínseca (factores XI y IX), y de forma más relevante al liberar de su transportador plasmático y activar el factor VIII, siendo el factor VIIIa un catalizador altamente eficiente del factor X. En conjunto, las vías mencionadas hasta el momento tienen como finalidad mantener e incrementar sustancialmente la producción de trombina, siendo un ambiente propicio para reclutar y activar más plaqueta.
Fase 3 o propagación: las plaquetas, una vez activadas, liberan sustancias quimioatrayentes como el difosfato de adenosina (ADP) y el tromboxano A2, además de cambiar su conformación física a una conformación que facilita la creación del tapón hemostático y expresar en su superficie fosfolípidos de carga negativa como la fosfatidilserina, los cuales en conjunto con el calcio sirven como templete para la formación de los complejos tenasa (factor VIIIa y IXa) y protrombinasa (factor Xa y Va), que tienen la capacidad de generar grandes cantidades de trombina. Finalmente, la trombina condiciona la polimerización de fragmentos de fibrinógeno en fibras de fibrina que darán estabilidad al coágulo inicial. Adicionalmente, la trombina activa al factor XIII, que estabilizará a la fibrina y brindará mayor resistencia al proceso de lisis (Figura 1) (9,12,13).
Fisiopatología de la hemostasia durante la insuficiencia hepática
Alteraciones en la hemostasia primaria
Desde etapas tempranas, en la enfermedad hepática existe una acumulación masiva de lípidos en el hepatocito que inducen la liberación de diversos mediadores inflamatorios, entre los que se pueden mencionar la interleucina (IL) 1 y 6, el factor de necrosis tumoral (TNF) y la estimulación del receptor LPS/Toll-like (TLR-4) presentes en las células de Kupffer 14,15. Estas vías desencadenan una disregulación del endotelio mediante la activación de la dimetilarginina asimétrica (ADMA), un inhibidor endógeno de la óxido nítrico sintetasa (eNOS) 16,17. Gracias a modelos animales, ahora se sabe que la disminución de la actividad de la eNOS es parte fundamental de la fisiopatología de la hipertensión portal 18,19.
Las plaquetas, además de servir como elemento celular en el tapón hemostático, poseen gránulos intracelulares con elementos capaces de modificar los procesos inflamatorios e inmunológicos dentro del hígado 10. Los gránulos α contienen factores de crecimiento cuya diana son las vías Akt (fosfatidilinositol-3-kinasa/AKT [PI3K-Akt]) y extracellular signal-regulated kinase (ERK) 1/2, ambas trascendentales para el hepatocito 20 al grado que recientes investigaciones sugieren que la transfusión de plaquetas puede mejorar la regeneración hepática 20,21. Los gránulos densos contienen nucleótidos de adenina que inactivan a las células estelares del espacio intersinusoidal y, en consecuencia, frenan los mecanismos profibróticos 21,22.
La trombocitopenia es una de las manifestaciones más frecuentes de la enfermedad hepática. Alrededor de un 76%-85% de los pacientes desarrolla algún grado de trombocitopenia (<150 x 103/µL), aunque solo un 13% presenta cifras <50 x 103//µL 23. Cabe recordar que el principal estímulo para la formación de plaquetas es la trombopoyetina (TPO), hormona producida primordialmente por los hepatocitos 24. La unión de la TPO con su receptor c-Mpl promueve las cascadas de activación de la quinasa de Janus 2 (JAK2) y la tirosina quinasa 2 (TYK2), además de hacer sinergia con otros estimulantes como la IL-3, IL-11 y el factor de célula progenitora (stem-cell factor), resultando en la proliferación y maduración de los megacariocitos 24,25. Panasiuk y colaboradores demostraron que en pacientes con cirrosis hepática existía una disminución de la producción de TPO y, en consecuencia, una disminución de plaquetas, así como una asociación directa entre la cuenta plaquetaria y el factor de crecimiento derivado de los hepatocitos (p <0,01) 26. Bajo este contexto se iniciaron ensayos clínicos sobre el uso de miméticos de la TPO en casos de falla hepática, especialmente en pacientes con hepatitis crónica viral. Al respecto, McHutchison y colaboradores evaluaron la adición del agonista del receptor de TPO (eltrombopag) a diferentes dosis (30, 50 y 75 mg) contra placebo durante 4 semanas en portadores de VHC que no podían iniciar terapia antiviral por cursar con trombocitopenia severa. La mejor respuesta se observó en el grupo de eltrombopag a 75 mg, en el que el 95% de pacientes elevó significativamente su cifra de plaquetas a niveles aceptables 27; Afdhal y colaboradores obtuvieron resultados favorables similares en las cohortes endothelin antagonist bosentan for lowering cardiac events (ENABLE) 1 y 2 con 9 semanas de seguimiento 28. Este mismo autor reportó en el estudio early levosimendan vs usual care in advanced chronic heart failure (ELEVATE) los beneficios de emplear eltrombopag para la elevación de la cifra de plaquetas antes de la realización de procedimientos invasivos (75 mg durante 14 días antes del procedimiento), lo que disminuye los requerimientos transfusionales en un 72%, aunque con un incremento de la trombosis de la vena porta en comparación con placebo 29.
Dentro de la fisiopatología de la trombocitopenia también se aprecia un incremento en la destrucción de plaquetas debido a hiperesplenismo o mediado por anticuerpos. Para diferenciar entre ambos mecanismos, en el primero se aprecia una trombocitopenia moderada acompañada también de una disminución de leucocitos por secuestro esplénico 30,31. Al respecto existen 2 grandes series que señalan a la esplenectomía (abordaje tradicional o laparoscópico) como medida terapéutica que ayuda a elevar las cifras de plaquetas (>100 x103/µL) y leucocitos en pacientes con cirrosis hepática 32,33. En modelos animales de fibrosis hepática se ha demostrado que la esplectomía reduce los niveles de factor de crecimiento transformante β (TGF-β), por esto se logra limitar e incluso reducir las áreas de cirrosis, además de cambios en el perfil inmunológico (incremento de linfocitos CD8+) 34,35.
Alteraciones en la hemostasia secundaria
La mayor parte de los factores procoagulantes como el fibrinógeno, la protrombina y los factores V, VII, IX, X, XI y XII son de síntesis hepática, por lo que sus niveles séricos se modifican en relación con el grado de daño hepático 36,37. Los factores V y VIII son los primeros en disminuir por poseer las vidas medias más cortas (12 y 4-6 horas, respectivamente) 38. Es común que los pacientes cirróticos sufran reducciones moderadas del factor VII, identificándose una relación significativa entre los niveles de factor VII y el tiempo de protrombina (TP) (r = 0,76, p <0,001) 39.
Cuando se evalúan las pruebas de coagulación en pacientes con hepatopatías, es importante descartar defectos congénitos; el más frecuente es la deficiencia de factor VII (1 caso por cada 500 000 nacimientos) 40. Esta deficiencia congénita se ha descrito en varias entidades clínicas (embarazo, cardiopatía, enfermedad pulmonar obstructiva, entre otras), aunque se tienen registrados pocos casos en pacientes cirróticos 41.
Trombosis e insuficiencia hepática
Previamente se mencionaron en detalle las vías moleculares de la trombina, y se mencionó su capacidad para generar estados de microtrombosis y consecuentes estados de isquemia en los sinusoides, lo que resulta en la expresión de factores profibrogénicos, que a su vez acelera y perpetúa el daño al parénquima hepático 42. Con este contexto no es raro que los pacientes con insuficiencia hepática, a pesar de ser proclives a eventos hemorrágicos, sufran de un estado de trombofilia altamente localizado en el sistema venoso esplácnico, cuya traducción clínica es alguna de las siguientes entidades: síndrome de Budd-Chiari (SBC), trombosis mesentérica (1 caso por millón de personas) y trombosis extrahepática de la vena porta (4 casos por millón de personas) 43; cabe recordar que hasta el 40% de los pacientes con cirrosis hepática sufre de trombosis de la vena porta 19,44,45,46. Algunos expertos sugieren que la reducción de velocidad de flujo en la vena porta es un hallazgo predictor de eventos trombóticos 45, por lo que una vez detectado debe iniciarse alguna estrategia antitrombótica, entre las que se pueden citar: terapia anticoagulante, angioplastia o colocación de endoprótesis vascular (stent), realización de una derivación transyugular intrahepática o incluso considerar trasplante hepático 47,48.
Factor de von Willebrand (FvW)
El FvW es una glucoproteína almacenada en los cuerpos de Weibel-Palade al interior de las células endoteliales y, una vez secretado, el FvW favorece la adhesión plaquetaria, además de unirse al factor VIII para evitar su pronta degradación 49. Durante la progresión del daño hepático existe una disminución de factores de coagulación, pero contrario a lo pensado también existe una elevación paralela del FvW y del factor VIII 50; el mecanismo subyacente parece ser la mayor actividad endotelial, el lugar de síntesis del FvW y el sitio donde se ubican las células involucradas en los procesos de fibrosis hepática que desembocarán en la reducción de los niveles de proteína C, S (factores anticoagulantes) y de la metaloproteasa ADAMTS13 (a disintegrin and metalloproteinasa with thrombospondin type 1 motif n.° 13) 44. Esta última es una proteasa sintetizada por las células estelares y es la encargada de fraccionar el FvW en pequeños multímeros capaces de unirse al factor VIII 51,52; la disminución de su actividad (<10%) causada por mutaciones, autoanticuerpos o por una producción reducida (insuficiencia hepática) da como resultado la producción de multímeros de FvW ultralargos (UL-FvW) que no pueden unirse al factor VIII, esto resulta en el incremento de factor VIII en su forma libre y activa 52,53. Un grupo de investigadores de la universidad de Nara, en Japón, han evidenciado que, en pacientes con insuficiencia hepática por alcohol, la actividad de ADAMTS13 disminuye y los niveles séricos de UL-FvW se incrementan; pero más importante aún es que estos hechos se correlacionan con el grado de severidad del daño hepático, e incluso plantean la posibilidad de emplearse como marcadores pronósticos 53,54.
Proteína C y S
La proteína C es uno de los principales anticoagulantes producidos en el hígado como un cimógeno que se activa en presencia del complejo trombina-trombomodulina sobre las superficies endoteliales. Una vez activada (proteína C activa, PCA) inhibe a 2 de los principales cofactores de la coagulación: factor Va y VIIIa 55.
La proteína S, codificada por el gen PROS1 y producida por los hepatocitos, células endoteliales, megacariocitos y osteoblastos, es parte fundamental no solo de la coagulación sino también de la ateroesclerosis, angiogénesis y la progresión de células neoplásicas 56. Es dependiente de la vitamina K y actúa estimulando la producción de PCA para regular la coagulación 57.
Durante el estudio de pacientes con eventos trombóticos debe tenerse en cuenta la disminución y falta de activación de estas proteínas debido al daño hepático, pero también descartarse otras situaciones clínicas concomitantes como resistencia a la PCA, defectos hereditarios de la coagulación (deficiencia de proteína C, S, factor V, Leiden, Factor II, mutaciones en metilentetrahidrofolato [MTHF] reductasa o hiperhomocistinemia) síndrome de antifosfolípidos e inclusive neoplasias 57,58; esto último en relación con el hecho de que existen homólogos de la proteína S en algunos tumores hepáticos 59,60.
Valoración de la hemostasia en pacientes hepatópatas
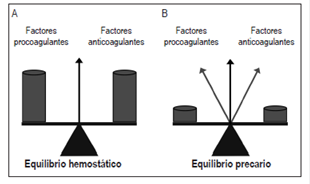
La hemostasia es una balanza que equilibra el gran peso de los factores procoagulantes y anticoagulantes; ante la deficiencia o incremento de alguno de estos, la balanza se moverá únicamente a favor del lado dominante. En pacientes con enfermedad hepática, esta balanza tendrá un precario equilibrio por la disminución de factores en ambos lados, el más mínimo aumento o disminución de alguno de los elementos ocasionará movimientos erráticos de la aguja de la balanza hasta que encuentre y marque el sitio dominante (Figura 2) 23,36,61. Se debe tener en cuenta esta comparación al momento de comenzar la valoración del estado hemostático en pacientes con enfermedad hepática, que será realizada al inicio con pruebas rutinarias (como la biometría hemática) y pruebas de coagulación básicas (TT: tiempo de trombina; TP: tiempo de protrombina; INR: international normalized ratio; TTPa: tiempo de tromboplastina parcial activado). Estas pruebas permiten evaluar únicamente los factores procoagulantes 23,36 y, salvo que se tenga una fuerte sospecha de algún defecto congénito, se puede complementar la evaluación con determinaciones de los factores de la coagulación 40,61.
Es importante decir que el TP y TTPa son pobres predictores de sangrado en personas hepatópatas; es decir, una cifra dentro del rango habitual no garantiza protección contra sangrado, ni cifras elevadas son sinónimo de sangrados incontrolables que ameriten medidas terapéuticas correctivas o preventivas 61,62. Aún se continúa en la búsqueda del método que permita determinar con fiabilidad el estado hemostático real de los pacientes con falla hepática, algunos de los más prometedores hasta el momento son el ratio de la generación de trombina y la tromboelastografía, aunque estas distan mucho aún de volverse pruebas rutinarias 61,62.
Vitamina k
La vitamina K (de la palabra alemana koagulationsvitamin, vitamina coagulante) está compuesta por un grupo de moléculas liposolubles derivadas de la 2-metil-1,4-naftoquinona 63; la filoquinona, de origen vegetal, se conoce como vitamina K1; la menaquinona, de origen bacteriano, se denomina vitamina K2; y las formas solubles en agua son la menadiona (vitamina K3) y el menadiol (vitamina K4) 63,64.
La vitamina K, al ser ingerida por vía oral, se absorbe en el intestino, pasa a la linfa y, ulteriormente, al torrente sanguíneo, donde se transporta del 75% al 90% hasta el hígado por diversas lipoproteínas de alta densidad, principalmente la apolipoproteína E (ApoE) (Figura 3). Este complejo no solo sirve de transporte, también facilita la unión e internalización desde el espacio de Disse hasta el interior del hepatocito 65. La captación rápida de filoquinona en el hígado se evidencia mediante la excreción de sus metabolitos en las sales biliares (30%-40%); otras vías de excreción son la orina (15%) y heces 65,66.
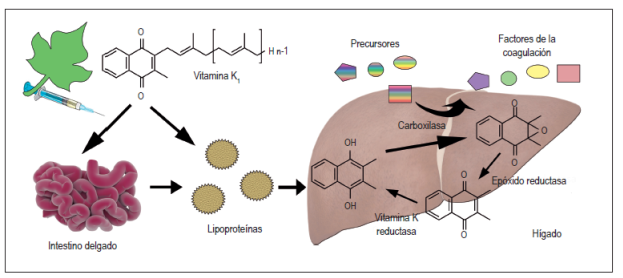
En hemostasia, la vitamina K cataliza la carboxilación de los factores de coagulación de síntesis hepática (VII, IX, X y protrombina) formando sitios de unión con el calcio en los residuos de ácido glutámico a los lados de la cadena proteica. Las proteínas C y S, anticoagulantes naturales, también requieren de vitamina K para su actividad 66,67. Debido a esto, es fácil comprender que los pacientes con hepatopatías crónicas importantes no responden adecuadamente a la suplementación con vitamina K y las alteraciones de la coagulación se vuelven de difícil control.
Administración de vitamina K
La principal indicación para la administración de vitamina K es la deficiencia de la misma en el recién nacido, alteración poco frecuente (0,1% de todos los recién nacidos vivos), pero potencialmente letal 65 y puede prevenirse con la administración de 2 mg vía oral en todos los recién nacidos 68,69. La segunda indicación terapéutica es como antídoto en pacientes bajo un régimen de anticoagulación con antagonistas de vitamina K y que sufren de hemorragia activa o alto riesgo de sangrado. Usualmente, se emplea vitamina K1 a una dosis media de 10-20 mg intravenosa (IV) o intramuscular (IM) (administración lenta, en un lapso no menor de 30 segundos) en conjunto con plasma fresco congelado o concentrados protrombínicos 70,71, medida que habitualmente en un lapso no mayor de 2 horas permite reducir el INR a niveles habituales; en casos severos, una segunda dosis similar puede administrarse en 8-12 horas 66,72.
Prescribir vitamina K para revertir las alteraciones en la hemostasia de los pacientes con daño hepático es una práctica constante por diversos especialistas bajo la hipótesis de que existe una deficiencia de factores de la coagulación dependientes de la vitamina K 73; sin embargo, es importante puntualizar que la mayor parte de los episodios hemorrágicos no los ocasiona una deficiencia en la hemostasia secundaria, sino más bien la ruptura de várices esofágicas o úlceras gástricas secundarias a hipertensión portal por el estado de trombofilia localizado 74. De acuerdo con la revisión sistemática de Martí-Carvajal y colaboradores, no hay ningún beneficio de la administración de vitamina K en pacientes con insuficiencia hepática y hemorragia, aunque esta afirmación se basa en series de casos o estudios observacionales, faltando aún el sustento de ensayos clínicos aleatorizados 75.
Consideraciones finales
Conocer las últimas evidencias científicas sobre el proceso de sangrado-coagulación permite comprender mejor por qué los pacientes con falla hepática pueden desarrollar alteraciones hemostáticas desde estadios tempranos de la enfermedad hepática, razón por la que es indispensable valorar la hemostasia secundaria mediante el TP o el INR, datos que además son necesarios para aplicar las escalas pronósticas habituales en hepatópatas.
Sin embargo, ahora se sabe que el principal factor de riesgo asociado con la persistencia de hemorragia es la fibrinólisis; así que, ante un evento hemorrágico, se debe poner igual o mayor importancia al hecho de brindar un soporte transfusional adecuado, acción que ha demostrado impactar positivamente en el pronóstico a corto plazo. Así entonces, el soporte transfusional puede indicarse solo de manera profiláctica (pacientes sin hemorragia que serán sometidos a biopsia hepática u otros procedimientos invasores) o con fines terapéuticos (hemorragia digestiva); en este último caso, con mayor sustento fisiopatológico que la administración empírica de vitamina K. Antes de iniciar el soporte transfusional, es importante considerar las complicaciones inherentes a la transfusión (formación de aloanticuerpos) y alteraciones hemodinámicas propias de pacientes con falla hepática como la lesión pulmonar aguda asociada con la transfusión (TRALI) o la sobrecarga de volumen (es importante recordar que la transfusión de plasma y plaquetas son las principales causas de TRALI) 76,77. También se hizo mención de la aparente utilidad de fármacos miméticos de la TPO para abatir trombocitopenias y evitar el uso de soporte transfusional, aunque la evidencia aún es limitada, por lo que en este momento aún no puede recomendarse su uso generalizado.

