










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.32 no.4 Bogotá Oct./Dec. 2017
https://doi.org/10.22516/25007440.180
Review articles
Colombian Guidelines for Diagnosis of Lipase Acid Deficiency
1Hospital Universitario San Vicente Fundación, Medellín, Colombia
2Hospital Pablo Tobón Uribe, Medellín, Colombia
3Hospital Infantil Napoleón Franco Pareja, Universidad del Sinú, Cartagena, Colombia
4Clínica Las Américas, Medellín, Colombia
5Unidad de Gastroenterología Pediátrica del Cesar, Valledupar, Colombia
6Centro Médico Almirante Colón, Bogotá, Colombia
7Hospital Infantil Napoleón Franco Pareja. Departamento de Pediatría, Universidad de Cartagena, Cartagena, Colombia
8Hospital Universitario Fundación Santa Fe de Bogotá, Universidad de los Andes, Bogotá, Colombia
9Fundación Hospital de la Misericordia, Bogotá, Colombia
10Fundación Hospital de la Misericordia, Bogotá, Colombia
11Clínica la Misericordia, Barranquilla, Colombia
12Fundación Cardioinfantil, Bogotá, Colombia. Correspondencia: carolinariveran@gmail.com.
13Hospital San Juan de Sahagún. Sahagún, Córdoba, Colombia
14Hospital Universitario Fernando Troconis, Santa Marta, Magdalena, Colombia
15Unidad Maternoinfantil del Tolima, Hospital Federico Lleras Acosta, Ibagué, Tolima, Colombia
16Hospital Pablo Tobón Uribe, Medellín, Colombia
17Fundación Hospital de la Misericordia, Bogotá, Colombia
18Hospital Universitario Fundación Santa Fe de Bogotá, Bogotá, Colombia
19Fundación Valle del Lili, Cali, Colombia
Introduction:
Lysosomal acid lipase deficiency (LAL-D) is an inherited autosomal recessive entity that leads to the accumulation of cholesterol and triglyceride esters in the liver, spleen and other organs. The age of onset and rate of progression vary greatly, possibly explained by mutations of the LIPA gene. Clinical manifestations are the same as those of other hepatic, cardiovascular and metabolic pathologies which makes it difficult to recognize in clinical practice. Objective: The objectives of these guidelines is to help clinicians recognize the major groups at risk for LAL-D and to improve its diagnosis.
Methodology:
This document was designed as a consensus of experts in gastroenterology, hepatology, endocrinology, genetics, pathology and pediatrics. A review of the literature regarding clinical manifestations and tools for diagnosis of LAL-D was conducted and the nominal group technique was followed.
Results:
Diagnostic algorithms which facilitate suspicion and diagnosis of LAL-D were generated by consensus for each of the risk groups.
Conclusions:
This guide proposes algorithms for the diagnosis of LAL-D based on clinical consensus. The algorithms seek to optimize diagnosis for patients with this pathology.
Keywords: Acid lipase deficiency; cholesterol ester deposition disease; Wolman’s disease; dyslipidemia; hepatomegaly
Introducción:
la deficiencia de lipasa ácida lisosomal (LAL-D) es una entidad de herencia autosómica recesiva que lleva a la acumulación de esteres de colesterol y triglicéridos en el hígado, bazo y otros órganos. La edad de inicio y la tasa de progresión son muy variables, lo que posiblemente sea explicado por las mutaciones presentes en el gen LIPA. Las manifestaciones clínicas son las mismas que para otras patologías hepáticas, cardiovasculares y metabólicas, lo que hace difícil reconocerla en la práctica clínica.
Objetivo:
proveer una guía que permita a los clínicos reconocer los principales grupos de riesgo en los cuales se debe sospechar de LAL-D y mejorar su diagnóstico.
Metodología:
este documento se diseñó como un consenso de expertos en el cual participaron médicos especialistas en gastroenterología, hepatología, endocrinología, genética, patología y pediatría. Se realizó una revisión de la literatura acerca de las manifestaciones clínicas y de las herramientas para el diagnóstico de LAL-D y se siguió la metodología de técnica de grupo nominal.
Resultados:
se generaron algoritmos diagnósticos por consenso para cada uno de los grupos de riesgo, que facilitaran la sospecha y el diagnóstico de LAL-D.
Conclusiones:
esta guía propone algoritmos para el diagnóstico de LAL-D con base en el consenso clínico, que buscan optimizar la ruta diagnóstica en los pacientes con dicha patología.
Palabras clave: Deficiencia de lipasa ácida; enfermedad por depósito de esteres de colesterol; enfermedad de Wolman; dislipidemia; hepatomegalia
Introduction
Guidelines and expert consensuses are documents that aim to help doctors evaluate the benefits and risks of diagnostic and therapeutic procedures related to a specific pathology. These documents are not intended to replace medical criteria and autonomy but to be useful guides for clinical decision-making. This guide was the result of a consensus of 20 experts from several medical specialties.
Each group of specialists conducted an exhaustive search of the available evidence in the current medical literature in PubMed and LILACS, the most frequently used search engines. Then the group reviewed and synthesized the evidence which became the basis for the expert consensus. Each group prepared a document on a related theme which was sent via e-mail to all the experts for their knowledge, analysis and contributions. The experts then presented the best available evidence on the diagnostic criteria for LAL-D in each risk group at a face-to-face scientific conference. This was followed by a debate in which each participant was able to express her or his opinion. Once the discussion points were analyzed. Consensus was defined as agreement of more than 80% of the members. Once consensus was reached, a report was issued with the recommendations and the design of algorithms that defined the path for the diagnosis of this pathology for the risk groups under consideration. This was the responsibility of the consensus coordinators. This document was then provided to all participants and consensus was reached.
LAL-D (OMIM 278000) is an autosomal recessive disease caused by mutations in the LIPA gene that drastically reduce the activity of the lysosomal acid lipase enzyme (LAL, EC 3.1.1.13)1. This enzyme is responsible for breaking down cholesterol esters and triglycerides, so dysfunction leads to their accumulation in the liver, spleen, macrophages and other organs 2. The disease is characterized by microvesicular steatosis that produces liver failure, elevation of serum transaminases, accelerated atherosclerosis and premature death 3. Infantile presentation was described in 1956, and this disease was historically called Wolman’s disease 4. It is characterized by vomiting, diarrhea, failure to thrive and hepatosplenomegaly (HSM) with rapid deterioration of liver functioning and death at an average age of 3.7 months 5. The late onset form was known as cholesterol ester deposit disease and was first described in 1963 6. It has a broad clinical spectrum and can occur in infants, children or adults 7. It is usually underdiagnosed and presents with liver compromise, elevated transaminases and elevated serum levels of LDL (low density lipoprotein) cholesterol and triglycerides with normal or low levels of HDL cholesterol (high density lipoprotein). Early death is due to hepatic insufficiency or accelerated atherosclerosis secondary to chronic hyperlipidemia 2.
Incidence has been estimated from genetic studies at 1/40,000 while the approximate incidence of the most severe forms is 1/500,000 8,9,10. Given its nonspecific clinical presentation and the relatively low number of published cases, it is believed that this entity has a high index of underdiagnoses 11. For this reason, these guidelines have been developers to offer health-care personnel a series of recommendations to guide themselves in developing suspicion of LAL-D and the ability to correctly diagnose it.
This guide was developed by a committee of experts in pediatrics, pediatric gastroenterology, pediatric endocrinology, hepatology, pathology and genetics using nominal group technique (NGT). Based on their experience and a wide literature review, the experts debated and discussed their different points of view to arrive at a list of recommendations representing their collective judgment. The objective was to propose guidelines that can be used in clinical practice for identification of patients with this pathology in Colombia.
Physiopathology
LAL-D originates in mutations of the LIPA gene located at 10q23.2. It has 10 exons and an approximate length of 45 kilobases (kb) 1. This entity is autosomal recessive inheritance whose most common defect is associated with a mutation at the site of exon 8 (c.894G> A) which is known as E8SJM. This mutation is present in about half of the affected patients 12, To date, more than 40 mutations which lead to alterations of the enzyme’s function and which are related to this pathology have been described 2. The most drastic ones are nonsense and frameshift mutations which are usually associated with severely affected patients, while cases in children and adults are associated with mutations whose effects on the protein are not as dramatic 13.
The LAL enzyme plays a key role in the metabolism of lipids since it is responsible for hydrolysis of cholesterol esters and triglycerides within lysosomes 14. The action of this enzyme generates free cholesterol which is involved in negative regulation of LDL receptors, in the inhibition of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase which is a rate controlling enzyme, and in the stimulation of acyl-coenzyme A (CoA):cholesterol acyltransferase (ACAT). Congenital deficiencies of this enzyme lead to lysosomal accumulation of cholesterol and triglyceride esters and to failure of the LDL receptor pathway. Therefore, in addition to this massive lysosomal accumulation of cholesterol esters, there is a systemic increase in synthesis of cholesterol 15.
Clinical Picture
Acid lipase deficiencies have a wide range of phenotypic presentations which manifest in very early childhood, in later childhood or in later stages of life.
Presentation in Infants
LAL-D usually manifests with the most acute symptoms when it manifests in the first days of life. Gastrointestinal symptoms include vomiting, diarrhea, steatorrhea, and abdominal distension secondary to hepatosplenomegaly. In addition, infants fail to thrive. 10 Increases of lipid deposits in the gastrointestinal tract cause thickening of the intestines resulting in malnutrition and malabsorption syndrome 16. Calcification of the adrenal glands is characteristic of this pathology, is present in up to 78% of affected children, and can cause cortical adrenal insufficiency 17.
Patients progress to multiorgan failure, especially to liver failure with cirrhosis and cachexia, and this leads to early an death: on average, at 3.7 months 5.
Presentation in Children and Adults
Presentation can be similar to that in infants. One third of patients experience severe gastrointestinal symptoms including frequent diarrhea, vomiting, abdominal pain, malabsorption and steatorrhea 2,14. Cholestasis, growth failure and gallbladder dysfunction are also important symptoms. 2 Classically, patients develop dyslipidemia and their total cholesterol increases as LDL and apolipoprotein B levels become elevated while HDL levels fall 2,14. This hyperlipidemia is associated with atherosclerosis, coronary artery disease and early catastrophic vascular events 2,18. Occasionally cholesterol deposits may be visible as xanthelasma develops (particularly in the palpebral region) 19.
Hepatomegaly with or without splenomegaly is common and is present in 99% of children and 74% of adults with LAL-D 2. Usually, organomegaly may be the first finding and is present many years before the diagnosis is made 20. Patients with splenomegaly may develop anemia or thrombocytopenia secondary to hypersplenism.
Liver involvement is common and elevated levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) may be early signs of liver damage. This can manifest with or without jaundice, steatosis, fibrosis or cirrhosis. Liver damage can lead to esophageal varices and progress to liver failure, cirrhosis, or hepatocellular carcinoma 21.
Lipid deposits in the intestines can lead to diarrhea and weight loss 22.
Diagnosis
LAL-D can be diagnosed by measuring the activity of the acid lipase enzyme or by determining mutations in the LIPA gene 14.
Measurement of LAL activity is done with dried blood spot testing (DBS) using a fluorometric substrate of 4-methylumbelliferyl palmitate (4 mU palmitate)(23). Because the presence of other lipases in the blood can interfere with measurement, Lalistat 2 (Chemical Tools, South Bend, IN, USA), a LAL inhibitor, is used. 23 The activity of LAL is determined by comparing total lipase activity with lipase activity in the presence of the inhibitor. The difference between the two can be attributed to the LAL enzyme. This measurement can be performed with leukocytes and with DBS, both of which have similar values for sensitivity and specificity 24. Nevertheless, measurement with DBS has had excellent results in discriminating between healthy and affected patients with intermediate values for carriers. It also has the advantages of being easy to take and transport and is very stable in the long term 23,24.
Molecular study through complete sequencing of the coding region of the LIPA gene allows identification of the causative mutations of the disease. The majority of patients are homozygous or have compound heterozygotes. However, some patients may have intronic mutations that are not always detected by sequencing 14. For this reason, the method of choice for diagnosis is measurement of enzymatic activity while molecular tests are considered to be complementary.
Figure 1 summarizes the algorithm for diagnosis of LAL-D.
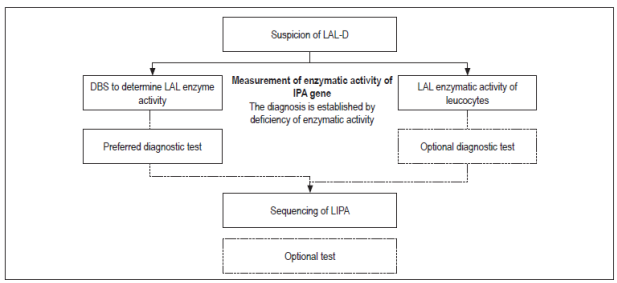
Other tests can be used when there is suspicion of this pathology, but they do not confirm the diagnosis.
Typically, liver biopsies will show microvesicular or mixed steatosis, but these conditions are not exclusive to LAL-D. Other findings including the presence of sea-blue histiocytosis, increased size of Kupffer cells with enlarged vacuoles, lipid drops and cholesterol crystals can support the diagnosis 9. Immunohistochemistry testing for cathepsin D and lysosomal-associated membrane proteins 1and 2 (LAMP1 and LAMP2), which are all lysosomal markers, can also be used 25.
Definition of Risk Groups
Because the clinical picture of LAL-D is similar to those of other cardiovascular, liver and metabolic diseases, it is possible to make inappropriate diagnoses that delay appropriate management of these patients.
Taking this into account, it was decided to establish an algorithm for each risk group to allow establishment of the appropriate stage of the diagnostic process for performing the LAL enzyme activity test. The theoretical framework and the diagnostic routes that have been developed are presented below.
Liver Diseases
Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) requires imaging or histological evidence of hepatic steatosis, and elimination of other possible causes for accumulation of liver fat such as alcohol consumption, the use of steatogenic medications and hereditary disorders. In most cases it is associated with metabolic risk factors such as obesity, diabetes mellitus (DM) and dyslipidemia. One special type of NAFLD is non-alcoholic steatohepatitis (NASH). NAFLD is defined as the presence of hepatic steatosis without evidence of hepatocellular damage. NASH is described as the presence of hepatic steatosis, inflammation, and damage to hepatocytes with or without fibrosis. 26
Common causes of hepatic steatosis include significant alcohol consumption, hepatitis C, cystic fibrosis, medications, parenteral nutrition, autoimmune hepatitis, severe malnutrition and medications including ethanol, corticosteroids, and estrogens. These should be considered and ruled out in the initial study of the patient, and a complete viral/immunological profile should be made to exclude the most common disorders.
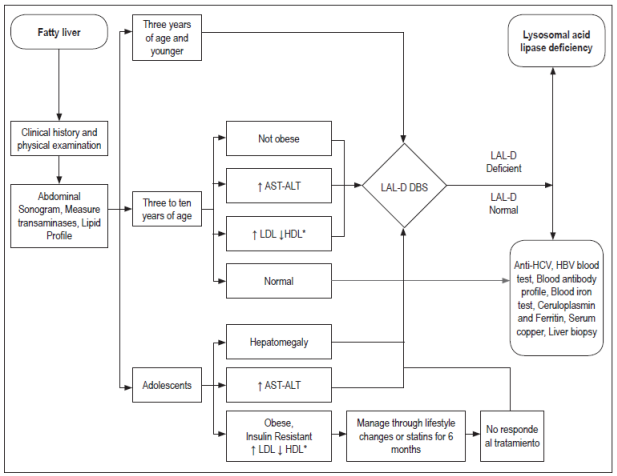
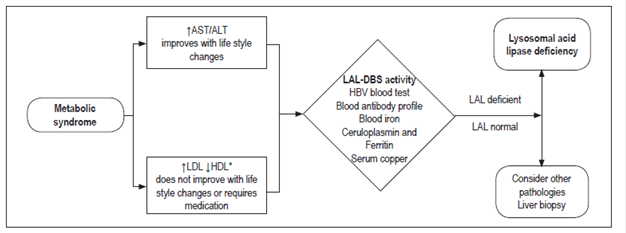
LAL-D should be suspected in patients with NASH or NAFLD who are have the following signs and symptoms:
Persistently elevated transaminases despite weight loss.
Dyslipidemia indicated by increased LDL values and decreased HDL according to age which does not improve after changes in lifestyle or medication.
Rapid progression of disease.
Figure 2 shows the algorithm for diagnosis of LAL-D in patients with fatty liver disease.
Hepatomegaly
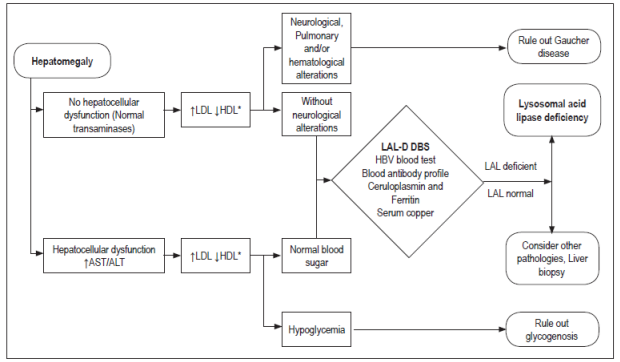
Normal liver size is based on values established by percussion, of extension below the right costal margin, or by the length of the vertical axis estimated by imaging studies. In general, it is estimated that measurements over 3.5 cm in neonates and 2 cm in children obtained by palpation under the right costal margin on the mid-clavicle line indicate hepatomegaly. 27
Hepatomegaly can be caused by five different mechanisms: inflammation, excessive deposition, infiltration, congestion and obstruction.
Inflammation is usually secondary to infections, toxins, radiation, autoimmune diseases and/or Kupffer cell hyperplasia that lead to inflammatory-type hepatomegaly.
Deposits are mainly glycogen, lipids, fat, metals and abnormal proteins.
Infiltration develops in cases of tumors, parasitic cysts and extramedullary hematopoiesis.
Vascular congestion is due to obstruction of venous drainage between the liver and the right atrium and may be intrahepatic or extrahepatic.
Biliary obstruction occurs in biliary atresia, choledochal cysts, cholelithiasis and tumors of the liver, bile duct, pancreas and duodenum. 27
A clinical history, physical examination, and complementary tests including a complete blood count, coagulation test, liver function tests, blood gas analysis, urinary sediment test and ultrasound are necessary for pediatric patients with hepatomegaly allow selection of other complementary tests to be performed.
Figures 3 and 4 show diagnostic algorithms for transaminases.
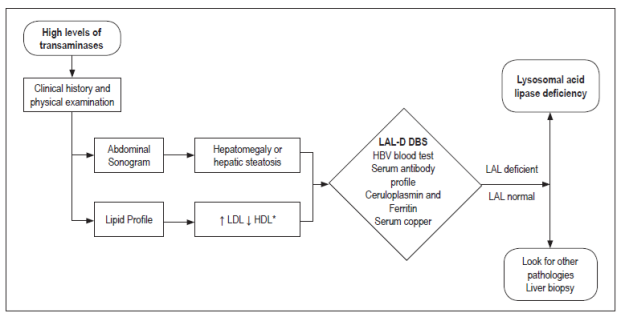
Figure 3 Algorithm for the suspicion of LAL-D in the presence of elevated transaminases. * Pediatric: LDL (mg/dL) ≥130 or HDL (mg/dL) ≤45; Adult: LDL (mg/dL) ≥160 or HDL (mg/dL) ≤40 (men)/≤50 (women). Ac: antibodies; Anti-HCV: hepatitis C virus antibodies; HBV: hepatitis B virus
Metabolic Hepatopathies
Gaucher Disease
Gaucher disease is an autosomal recessive disorder caused by deficiency of glucocerebrosidase (GCase), a lysosomal hydrolase. Its estimated incidence is 1:200,000.
The disease should be suspected when histopathology of biopsies from the bone marrow, liver and/or spleen reveal Gaucher cells, but measurement of GCase activity in leukocytes or fibroblasts obtained from a skin biopsy is the gold standard for diagnosis 28. A molecular study of the GBA gene can be carried out for confirmation.
Clinically, this disease affects a number of systems, so its phenotypes are divided into non-neuronopathic and neuronopathic. Clinical manifestations of non-neuronopathic Gaucher disease vary greatly, but most are hematological including anemia and thrombocytopenia, visceral including hepatosplenomegaly, or compromise bones. Secondary neurological complications may occur as a result of nerve compression of the spinal cord due to vertebral collapse 28-30.
Acid Sphingomyelinase Deficiency (Niemann-Pick Disease A and B)
Acid sphingomyelinase (ASM) deficiency is an autosomal recessive disorder caused when enzymatic activity of ASM is less than 10% of control sample activity or by the presence of pathogenic biallelic mutations in the SMPD1 gene (also called the ASM gene) 31,32.
Classically, this disorder has been divided into neuronopathic (Niemann-Pick A disease) or non-neuronopathic (Niemann-Pick B disease) (Table 1). However, there are intermediate forms between both phenotypes 33.
Glycogenosis
Glycogen is a polysaccharide formed by glucose molecules linked together primarily by alpha 1,4 (α-1,4) and with 7% to 10% alpha 1,6 (α-1,6) bonds. In the liver, glycogen has the mission of maintaining blood sugar levels while in muscles it is used to obtain energy (ATP) during muscle contractions 34,35. Genetic disorders that affect the glycogen synthesis and degradation pathways and glycogen use are included under this term.
Genetic disorders that affect glycogen metabolism can be divided into two categories on the basis of manifestations, diagnostic criteria and treatment: those with hypoglycemic liver pathophysiology and those which are muscular. Glycogenosis types Ia, Ib, IIIa, IIIb and VI are in the first group while glycogenosis types V, VII and glycolysis defects that do not cause glycogen accumulation are within the second group. However, some entities such as glycogenosis types II and IV have peculiar pathophysiologies 34,35. The presence of hyperlipidemias and hepatomegaly in these types of glycogenosis require that they be considered in differential diagnosis of LAL-D.
Figure 5 describes the diagnostic algorithm for metabolic liver diseases.
Metabolic Syndrome
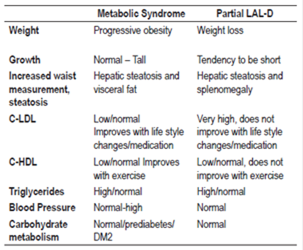
Metabolic Syndrome is a constellation of risk factors of interrelated metabolic origin that directly promote the development of arterial disease, diabetes mellitus type 2 (DM2), fatty liver disease and several cancers 36.
According to the International Diabetes Federation, the criteria for this syndrome have been defined as the presence of at least three of the following five factors: 37
Large waist size according to the criteria of the country and the specific population.
Triglycerides over 150 mg/dL.
Low HDL cholesterol (<40 mg/dL in men and <50 mg/dL in women).
High in blood pressure (systolic pressure ≥130 mm Hg or diastolic pressure ≥85 mm Hg).
High fasting glucose (blood glucose> 100 mg/dL).
Numerous contributing factors have been described for development of metabolic syndrome. They include genetic factors, lifestyle factors such as diet and physical activity, obesity and insulin resistance. All are important for pathogenesis of metabolic syndrome, but insulin resistance is a key factor that connects and contributes to the development of the disorder. High levels of fatty acids and an abnormal adipocytokine profile can establish insulin resistance and contribute to the pathogenesis of the disease. 38
Table 1 summarizes the differences between LAL-D and metabolic syndrome.
The expert consensus for diagnosis of patients who have signs and symptoms of metabolic syndrome is summarized in Figure 6.
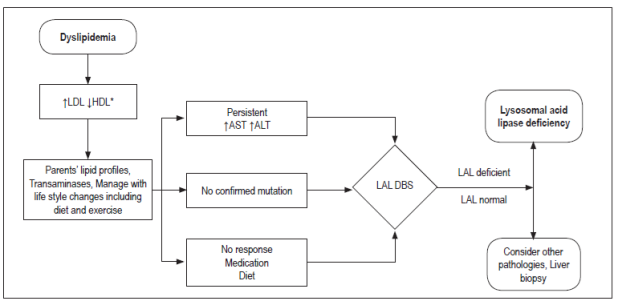
Dyslipidemias
Familial Hypercholesterolemia
Familial Hypercholesterolemia is a disease based on an autosomal dominant inheritance that leads to severe elevation of total cholesterol and LDL. There are homozygous and in heterozygous forms of presentation.
Homozygous patients present symptoms consistent with peripheral vascular disease, ischemic heart disease, cerebrovascular disease or aortic stenosis. They may also develop joint symptoms such as tendonitis and arthralgia, and at birth. At birth and in early childhood there may be cutaneous xanthomas 39. Most homozygous patients who do not receive treatment die before 30 years of age.
Heterozygous patients are usually asymptomatic, but they may have histories of severe hypercholesterolemia from childhood and may later develop xanthelasmas or xanthomas 39.
The fundamental finding for diagnosis of both homozygous and heterozygous forms is the cholesterol level in the absence of secondary causes of hypercholesterolemia. Nevertheless, the definitive diagnosis can only be made with molecular confirmation through analysis of the LDLR, APOB, or PCSK9 genes 40.
Key findings in the lipid profile are:
Homozygous: total cholesterol and LDL levels> 600 mg/dL and triglycerides within the reference range.
Heterozygous: LDL cholesterol levels> 250 mg/dL in children under 20 years of age and greater than 290-300 mg/dL in adults. Triglycerides are within the reference ranges 41.
Familial Combined Hyperlipidemia
Familial combined hyperlipidemia is the most frequent inherited lipid metabolism disorder that is associated with mixed hyperlipidemia and premature cardiovascular disease. Although symptoms may appear in childhood, it usually manifests in the second decade of life with hypercholesterolemia or hypertriglyceridemia. It is frequently associated with numerous metabolic abnormalities such as hypertension, insulin resistance, DM2, central obesity, hepatic steatosis and metabolic syndrome 42.
It is characterized by an autosomal dominant inheritance with high penetrance and oligogenic etiology.
The following criteria should be taken into account for diagnosis:
1. Affected person
In adults: total cholesterol > 240 mg/dl (or c-LDL> 160 mg/dL) and/or triglycerides > 200 mg/dL.
In children under 20 years of age : total cholesterol > 200 mg/dL (or c-LDL > 130 mg/dL) or triglycerides > 120 mg/dL.
Discard secondary causes: body mass index (BMI) > 35 kg/m2, glycosylated hemoglobin (HbA1c) > 10%, uncontrolled hypothyroidism or alcoholism (more than 40 g alcohol/day).
2. Affected family
Two or more first-degree members (parents, siblings or children) affected by mixed hyperlipidemia or combinations of pure hypercholesterolemia (IIa), mixed hyperlipidemia (IIb) and hypertriglyceridemia (IV).
Families with tendinous xanthomas or LDL-c levels > 300 mg/dL in two or more first-degree relatives with phenotype IIa are excluded.
Family history of premature atherosclerotic cardiovascular disease (prior to sixty years of age) 42.
Conclusion
LAL-D’s prevalence is small, but this may be due to large amounts of under-diagnosis and misdiagnosis. These guidelines aim at defining which patients should be tested for this disease, so that these patients will receive timely and effective treatment to change their prognoses and quality of life.
These guidelines for developing diagnostic suspicion are a reference for the clinical practices of general practitioners and specialists.
Referencias
1. Aslanidis C, Ries S, Fehringer P, et al. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33(1):85-93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8617513 [ Links ]
2. Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-43. http://dx.doi.org/10.1016/j.jhep.2013.02.014 [ Links ]
3. Sloan HR, Fredrickson DS. Enzyme deficiency in cholesteryl ester storage idisease. J Clin Invest. 1972;51(7):1923-6. [ Links ]
4. Abramov A, Schorr S, Wolman M. Generalized xanthomatosis with calcified adrenals. AMA J Dis Child. 1956;91(3):282-6. [ Links ]
5. Jones SA, Valayannopoulos V, Schneider E, et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 2016;18(5):452-8. Available from: http://www.nature.com/doifinder/10.1038/gim.2015.108 [ Links ]
6. Fredrickson D. Newly recognized disorders of cholesterol metabolism. Ann Intern Med. 1963;58:718. [ Links ]
7. Fasano T, Pisciotta L, Bocchi L, et al. Lysosomal lipase deficiency: molecular characterization of eleven patients with Wolman or cholesteryl ester storage disease. Mol Genet Metab. 2012;105(3):450-6. [ Links ]
8. Muntoni S, Wiebusch H, Jansen-Rust M, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27(8):1866-8. [ Links ]
9. Pisciotta L, Fresa R, Bellocchio A, et al. Cholesteryl Ester Storage Disease (CESD) due to novel mutations in the LIPA gene. Mol Genet Metab . 2009;97(2):143-8. http://dx.doi.org/10.1016/j.ymgme.2009.02.007 [ Links ]
10. Grabowski G, Charnas L, Du H. Lysosomal acid lipase deficiencies: the Wolman disease/cholesteryl ester storage disease spectrum. Online Metab Mol Bases Inherit Dis. 2012;1-26. [ Links ]
11. Freudenberg F, Bufler P, Ensenauer R, et al. Cholesteryl ester storage disease: an easily missed diagnosis in oligosymptomatic children. Z Gastroenterol. 2013;51(10):1184-7. [ Links ]
12. Scott SA, Liu B, Nazarenko I, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58(3):958-65. [ Links ]
13. Saito S, Ohno K, Suzuki T, et al. Structural bases of Wolman disease and cholesteryl ester storage disease. Mol Genet Metab . 2012;105(2):244-8. [ Links ]
14. Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30. [ Links ]
15. Zhang B, Porto AF. Cholesteryl ester storage disease: protean presentations of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr. 2013;56(6):682-5. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00005176-201306000-00020 [ Links ]
16. Nchimi A, Rausin L, Khamis J. Ultrasound appearance of bowel wall in Wolman’s disease. Pediatr Radiol. 2003;33(4):284-5. [ Links ]
17. Jones SA, Bernstein D, Bialer M, et al. Severe and rapid disease course in the natural history of infants with lysosomal acid lipase deficiency. Mol Genet Metab . 2014;111(2):S57-8. Available from: http://www.sciencedirect.com/science/article/pii/S109671921300557X%5Cnhttp://linkinghub.elsevier.com/retrieve/pii/S109671921300557X [ Links ]
18. Elleder M, Chlumská A, Hyánek J, et al. Subclinical course of cholesteryl ester storage disease in an adult with hypercholesterolemia, accelerated atherosclerosis, and liver cancer. J Hepatol . 2000;32(3):528-34. [ Links ]
19. Elleder M, Ledvinová J, Cieslar P, et al. Subclinical course of cholesterol ester storage disease (CESD) diagnosed in adulthood. Report on two cases with remarks on the nature of the liver storage process. Virchows Arch A Pathol Anat Histopathol. 1990;416(4):357-65. [ Links ]
20. Ameis D, Brockmann G, Knoblich R, et al. A 5’ splice-region mutation and a dinucleotide deletion in the lysosomal acid lipase gene in two patients with cholesteryl ester storage disease. J Lipid Res. 1995;36(2):241-50. [ Links ]
21. Riva S, Spada M, Sciveres M, et al. Hepatocarcinoma in a child with cholesterol ester storage disease. Dig Liver Dis. 2008;40(9):784. [ Links ]
22. Drebber U, Andersen M, Kasper HU, et al. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol. 2005;11(15):2364-6. [ Links ]
23. Hamilton J, Jones I, Srivastava R, et al. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413(15-16):1207-10. http://dx.doi.org/10.1016/j.cca.2012.03.019 [ Links ]
24. Civallero G, Michelin K, de Mari J, et al. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin Chim Acta . 2006;372(1-2):98-102. [ Links ]
25. Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60(7):1107-13. [ Links ]
26. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142(7):1592-609. http://dx.doi.org/10.1053/j.gastro.2012.04.001 [ Links ]
27. Wolf AD, Lavine JE. Hepatomegaly in neonates and children. Pediatr Rev. 2000;21(9):303-10. [ Links ]
28. Linari S, Castaman G. Clinical manifestations and management of Gaucher disease. Clin Cases Miner Bone Metab. 2015;12(2):157-64. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4625773&tool=pmcentrez&rendertype=abstract [ Links ]
29. vom Dahl S, Harzer K, Rolfs A, et al. Hepatosplenomegalic lipidosis: what unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase -1 exon 8 splice junction mutation. J Hepatol . 1999;31(4):741-6. [ Links ]
30. Masi L, Brandi ML. Gaucher disease: the role of the specialist on metabolic bone diseases. Clin Cases Miner Bone Metab . 2015;12(2):165-9. [ Links ]
31. Zampieri S, Filocamo M, Pianta A, et al. SMPD1 Mutation Update: Database and Comprehensive Analysis of Published and Novel Variants. Hum Mutat. 2016;37(2):139-47. [ Links ]
32. Rhein C, Mühle C, Kornhuber J, et al. Alleged Detrimental Mutations in the SMPD1 Gene in Patients with Niemann-Pick Disease. Int J Mol Sci. 2015;16(6):13649-52. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4490514&tool=pmcentrez&rendertype=abstract [ Links ]
33. McGovern MM, Wasserstein MP, Giugliani R, et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics. 2008;122(2):e341-9. [ Links ]
34. Bodamer OA, Feillet F, Lane RE, et al. Utilization of cornstarch in glycogen storage disease type Ia. Eur J Gastroenterol Hepatol. 2002;14(11):1251-6. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id= 12439121&retmode=ref&cmd=prlinks%5Cnpapers3://publication/uuid/CB712C8E-D63E-4AC0-9B42-4C5DF0A2230C [ Links ]
35. Avaria M, Kleinsteuber S. Trastorno del metabolismo de hidratos de carbono. En: Colombo M, Cornejo Espinoza V, Raimann Ballas EA (editoras). Errores innatos en el metabolismo del niño. 3.ª edición. Santiago: Editorial Universitaria; 2010. p. 458-66. [ Links ]
36. Grundy SM, Brewer HB Jr, Cleeman JI, et al. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109(3):433-8. [ Links ]
37. Alberti KGMM, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640-5. Available from: http://circ.ahajournals.org/content/120/16/1640.abstract%5Cnhttp://circ.ahajournals.org/content/120/16/1640.full.pdf [ Links ]
38. Weiss R, Bremer AA, Lustig RH. What is metabolic syndrome, and why are children getting it? Ann N Y Acad Sci. 2013;1281:123-40. [ Links ]
39. Varghese MJ. Familial hypercholesterolemia: A review. Ann Pediatr Cardiol. 2014;7(2):107-17. Available from: http://www.annalspc.com/text.asp?2014/7/2/107/132478 [ Links ]
40. Wiegman A, Gidding SS, Watts GF, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425-37. [ Links ]
41. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478-90a. [ Links ]
42. Mata P, Alonso R, Ruíz-García A, et al. Hiperlipidemia familiar combinada: documento de consenso. Aten Primaria. 2014;46(8):440-6. http://dx.doi.org/10.1016/j.aprim.2014.04.013 [ Links ]
Received: April 04, 2017; Accepted: October 06, 2017
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
