











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El cáncer colorrectal (CCR) se considera un problema de salud pública con una amplia distribución geográfica, según el registro de GLOBOCAN 2012 1. Actualmente, el CCR es el tercer cáncer más común en hombres y mujeres en el mundo, con una alta tasa de incidencia en países desarrollados 1; por el contrario, los países en vía de desarrollo tienen una baja incidencia 2. Sin embargo, datos recientes de la Agencia Internacional de Investigaciones del Cáncer (IARC) muestran un aumento en las tasas de incidencia y mortalidad en países menos desarrollados 3,4.
En Colombia, se reporta un aumento de las tasas de incidencia y mortalidad del CCR durante las últimas décadas 5,6. De acuerdo con las cifras de GLOBOCAN 2012 para Colombia, el CCR ocupa el cuarto lugar en incidencia y mortalidad en ambos sexos; al año se presentan 4107 nuevos casos, la mayoría diagnosticados en estados avanzados de la enfermedad 1.
El CCR ocurre de forma esporádica en cerca del 80% y el 20% restante tiene una historia familiar; las mutaciones germinales en los genes APC y MLH1 predisponen al CCR de tipo hereditario 7,8,9. El CCR se origina por diferentes alteraciones genéticas que están involucradas en el inicio y desarrollo de esta enfermedad 10; dichas alteraciones afectan la expresión de múltiples genes, promoviendo la trasformación de la mucosa normal del colon hacia un pólipo benigno, que progresa hacia un adenoma temprano, luego se vuelve intermedio hasta, finalmente, avanzar a un adenocarcinoma de colon 10.
Por una parte, se conocen diferentes vías moleculares para explicar el desarrollo del CCR. Inicialmente, se propuso el modelo clásico de progresión de adenoma hacia carcinoma por Fearon y Vogelstein 11 que se denomina vía tradicional o supresora, e involucra la inactivación de los genes supresores de tumores APC y TP53, y mutaciones en los oncogenes KRAS, DCC y SMAD 11. La segunda vía se denomina mutadora y se relaciona con mutaciones en los genes del sistema de reparación MMR, principalmente MLH1 y MSH2, que inducen inestabilidad microsatelital (MSI, microsatellite instability) en las células tumorales 12. La tercera vía implicada en el desarrollo del CCR es la epigenética, que consiste en la represión de la expresión génica mediante la metilación de la región promotora de los genes supresores tumorales o de reparación 13.
Por otra parte, el término epigenética se estableció en 1942 por Conrad Hal Waddington para describir los mecanismos que modifican la estructura de la cromática y afectan los niveles de la expresión de genes, sin cambios en la secuencia de ácido desoxirribonucleico (ADN); estos mecanismos incluyen la metilación, acetilación e hidroxilación del ADN, modificación de proteínas tipo histonas y la remodelación de la estructura de la cromatina; así como los ácidos ribonucleicos (ARN) no codificantes o micro-ARN. Las modificaciones epigenéticas se pueden inducir por factores externos e internos que podrían tener efectos similares a los de las mutaciones patogénicas, puesto que podrían inactivar la expresión de diversos genes en un determinado tejido; lo anterior se ha evidenciado en la carcinogénesis de diferentes órganos y tiene gran importancia en el desarrollo del cáncer, teniendo en cuenta que estas modificaciones afectan la expresión de genes supresores de tumores o genes del sistema de reparación del ADN 14.
El propósito de esta revisión es describir las principales modificaciones epigenéticas relacionadas con el CCR y su importancia en las aplicaciones clínicas que podrían ser utilizadas como biomarcadores para la detección temprana del CCR.
Modificación de las histonas
Un mecanismo por el que se induce un control transcripcional sobre la expresión de los genes involucra modificaciones en las proteínas histonas que están asociadas con el ADN 15,16. Se conoce que las modificaciones covalentes postraduccionales en regiones específicas de las histonas constituyen un mecanismo epigenético que regula y modifica la estructura de la cromatina; por lo anterior, se considera que dichas modificaciones podrían estar asociadas con el origen y progresión del cáncer 16.
El entendimiento de las modificaciones de las histonas en el desarrollo del cáncer en humanos hasta el presente es muy limitado. Las modificaciones en las histonas ocurren comúnmente en H2A, H2B, H3 y H4, las cuales hacen parte del octámero de histonas y están organizadas en una estructura cilíndrica (15,16. El nucleosoma es una unidad compuesta de aproximadamente 150 a 200 pares de bases de ADN que se disponen alrededor de las histonas nucleosómicas; la porción N-terminal de cada histona que emerge de esta estructura compacta es el blanco en las que ocurren las modificaciones postraduccionales. Los principales tipos de modificaciones son la fosfoliración, glucosilación, ADP-ribosilación, ubiquitinación, sumoilación y, las más comunes, acetilación y metilación (Figura 1) 15,16.
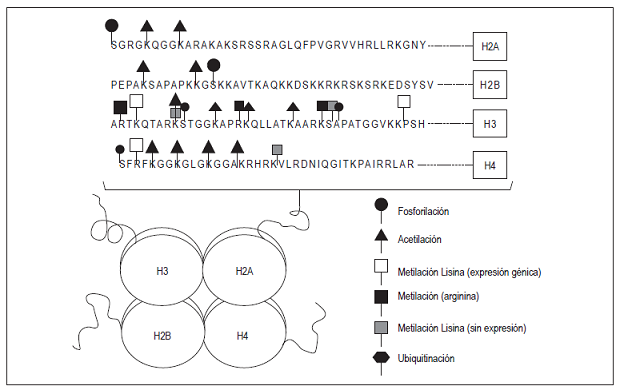
Figura 1 Principales modificaciones postraduccionales en los residuos N-terminales de las histonas. Las 4 proteínas histonas (H2A, H2B, H3 y H4) tienen una estructura cilíndrica denominada núcleo histónico. Las principales modificaciones covalentes de cada histona son la fosfoliración, acetilación, metilación y ubiquitinación. En la imagen se observa la secuencia de aminoácidos que componen el residuo N-terminal de cada histona y el sitio donde ocurre cada tipo de modificación.
La acetilación y la metilación son las modificaciones epigenéticas más estudiadas en el cáncer. Se ha observado que los residuos de aminoácidos, el tipo de modificaciones y dominio de la histona están asociados con el desarrollo y progresión del cáncer 16,17,18. En general, dentro de los mecanismos epigenéticos, las modificaciones de las histonas son de gran importancia y objeto de muchos estudios en la epigenética del cáncer.
Los estados de acetilación/desacetilación y metilación/desmetilación de los residuos de lisina y arginina en las histonas están entre las modificaciones más estudiadas y entendidas. En general, se conoce que la hipoacetilación reprime la expresión génica; mientras que la hiperacetilación de las histonas activa la transcripción de los genes. La acetilación en motivos específicos de las histonas desestabiliza la fibra de cromatina, lo que permite un aumento de la movilidad de los nucleosomas en los cromosomas y, en este estado, se unen los factores de transcripción al ADN 19.
Actualmente, se conoce que la desacetilación de la histona H3K9 se relaciona con la represión de la transcripción de la E-cadherina en células de tumores colorrectales; otros estudios informan que la pérdida de la expresión de la E-cadherina por metilación y la subsecuente pérdida de la adhesión celular parecen ser un paso crítico en la capacidad que adquieren las células tumorales para invadir tejidos adyacentes y llevar a cabo la metástasis. Diferentes estudios han demostrado que la expresión de esta proteína es nula o muy escasa en carcinomas poco diferenciados, incluidos los de CCR 20,21.
Metilación del ADN
La regulación de la expresión génica es el producto de la interacción entre los factores de inicio de la transcripción y las secuencias promotoras ubicadas antes del codón de inicio (ATG). En células normales, el 60% de los genes codificantes en humanos, la región promotora es rica en secuencias de citosina-guanina (C-G, denominadas islas CpG) 22.
Un mecanismo por el que la célula puede inhibir completamente la expresión génica es evitar la unión de los factores de transcripción a secuencias reguladoras del ADN mediante la metilación de las regiones promotoras, específicamente en las islas CpG, por la unión covalente de un grupo metilo (CH3) en el carbono 5´ de la citosina por medio de las enzimas ADN metiltransferasas (DNMT) (Figura 2); las islas CpG comprenden regiones entre 200 y 2000 pares de bases con una proporción de CG >50% 23. La metilación reprime la transcripción por la unión de proteínas ligadoras de metil-CpG (MBP) que interactúan con las secuencias CpG metiladas, lo que impide la unión de los factores de transcripción con secuencias de ADN; esto produce la inactivación los de genes 24,25. Es importante resaltar que las islas CpG que no están dentro de un gen activo se mantienen en su mayoría metiladas, mientras que las islas CpG que no se encuentran metiladas hacen parte de la región promotora de genes que se expresan constitutivamente (Figura 3) 10.
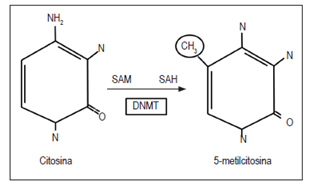
Figura 2 Metilación de la citosina en el carbono 5 mediante la enzima DNMT. Esta enzima cataliza la unión de un grupo metilo CH3 en el carbono 5 de la citosina, empleando como molécula donadora del grupo CH3 la S-adenosil metionina (SAH). SAH: S-adenosil L-homocisteína.
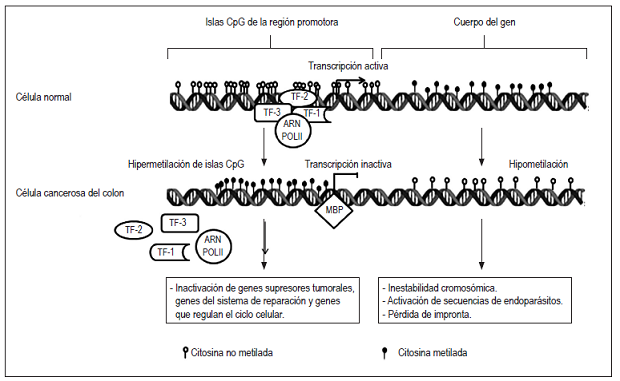
Figura 3 Estado de metilación del ADN. En la región promotora de una célula normal del colon, las islas CpG dentro del promotor no se encuentran hipermetiladas (círculos en blanco), lo cual permite que se una el ARN polimerasa II (ARN POLII) y los factores de transcripción, como el FT-2, FT-3 y FT-1; de esta manera, se da la transcripción normal del gen. En contraste, en una célula de adenocarcinoma de colon, las islas CpG de la región promotora se encuentra hipermetilada (círculos negros); lo que reprime la transcripción por la unión de proteínas MBP que interactúan con las secuencias CpG metiladas; a su vez, esto impide la unión de los factores de transcripción con el ADN y, por tanto, se produce la inactivación génica.
Entre las modificaciones que ocurren en el genoma, la metilación tiene funciones importantes como regular los procesos de replicación, transcripción, reparación del ADN y expresión génica, en general. Adicionalmente, esta modificación del ADN es necesaria para la inactivación permanente de determinadas regiones con genes que no se expresan después del desarrollo embrionario y para la inactivación del cromosoma X; también es esencial para la coordinación de procesos transcripcionales durante el desarrollo embrionario y la diferenciación celular (10.
Hipermetilación del adn en el cáncer colorrectal
La metilación del ADN se ha estudiado ampliamente en diferentes tipos de cáncer 19,26,27. Asimismo, es bien conocida la asociación entre la hipermetilación en las islas CpG localizadas cerca de la región promotora de genes y la inactivación de la transcripción génica, incluyendo la represión de la transcripción de genes supresores de tumores, genes que regulan el ciclo celular y genes del sistema de reparación del ADN 28. Estos genes usualmente están involucrados en múltiples procesos biológicos importantes como: la proliferación celular, apoptosis, angiogénesis, invasión y adhesión celular 29.
Particularmente en el CCR, múltiples estudios han informado que es frecuente la hipermetilación de la región promotora de los genes RB1, APC, MLH1, MGMT, CDH1, CDKN2A, RUNX3, y RASSF1A, entre muchos otros (Tabla 1) 26,30-39.
Tabla 1 Principales genes hipermetilados en CCR. Se presenta una lista de diversos genes frecuentemente metilados en CCR, la función y la técnica empleada del análisis epigenético.
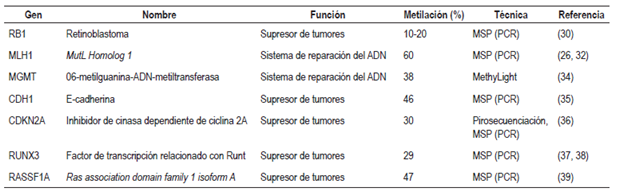
MSP: methylation-specific PCR; PCR: reacción en cadena de la polimerasa.
Por una parte, uno de los primeros descubrimientos del efecto de la metilación en las islas CpG en un gen supresor de tumores en cáncer fue el del retinoblastoma (RB1), que se encuentra hipermetilado entre el 10%-20% de los casos de CCR. El RB1 codifica para una nucleoproteína que tiene una función importante en la regulación del ciclo celular; la pérdida de la función de este gen causa una proliferación celular incontrolada; este gen se encuentra alterado en una gran variedad de tumores, incluido el CCR 30.
Por otra parte, la hipermetilación del promotor del gen MLH1 se relaciona con el desarrollo de CCR esporádico y hereditario que presenta MSI 26. El gen MLH1 hace parte del sistema de reparación de bases mal apareadas (MMR, mismatch repair); los defectos en estos genes por mutaciones puntuales o por el silenciamiento epigenético son la causa de la MSI en el desarrollo del CCR 33. Este gen se encuentra metilado hasta en el 60% de los casos esporádicos de CCR 32. En algunos casos, se ha observado que el gen MLH1 está metilado en tejido adyacente al tumor con una apariencia morfológica normal, por lo que se propone como un biomarcador para la detección temprana del CCR; sin embargo, se requieren más estudios para establecer lo anterior 31,32.
De manera similar, el gen MGMT, un gen del sistema de reparación del ADN encargado de remover aductos del ADN y prevenir la formación de MMR, se encuentra hipermetilado en el 38% de los pólipos colorrectales; en estos casos, se asocia la inactivación de este gen con el desarrollo CCR y con mutaciones en otros genes involucrados en este tipo de cáncer como APC, KRAS Y TP53 34.
Asimismo, estudios de metilación en el gen supresor de tumores CDH1 han mostrado que este gen está hipermetilado en el 46% de los casos de CCR 35; como consecuencia de esta modificación epigenética, se disminuyen la expresión y la función de la proteína E-cadherina, favoreciendo de esta manera un aumento en la proliferación celular, la invasión y la metástasis de las células tumorales de colon. También la inactivación de este gen podría asociarse con alteraciones de otros genes importantes en el desarrollo de CCR, como es el caso del gen APC 35.
Por su parte, el gen CDKN2A (p16), otro supresor de tumores, se encuentra hipermetilado en el 30% de los casos con CCR. La represión transcripcional de este gen también favorece el desarrollo, progresión e invasión de las células transformadas en este tipo de cáncer. Adicionalmente, esta modificación se encuentra con frecuencia en el colon ascendente y se asocia con un mal pronóstico en los pacientes con esta enfermedad 36.
Otra de las evidencias de la inactivación epigenética en CCR ocurre con el gen RUNX3, que actúa como supresor de tumores en el epitelio del colon. Su inactivación se presenta aproximadamente en el 20% de los casos de CCR 37. De estos resultados, se propone una asociación significativa entre la inactivación de RUNX3 y CCR con MSI positiva; se concluye que el gen RUNX3 es importante en el desarrollo de CCR 37,38.
El gen RASSF1A se encuentra comúnmente hipermetilado en el CCR, es un gen supresor de tumores que actúa en la progresión de la fase G1 hacia la S del ciclo celular 39,40. La metilación de este gen se observa en el 47% de los casos de CCR, con un estado avanzado de la enfermedad, con metástasis e invasión linfática. Asimismo, se propone una correlación significativa entre los casos de CCR con mutación en el codón 12 de gen KRAS y el estado de hipermetilación de este gen, por lo que se sugiere que, además de considerarse importante en el desarrollo del CCR, también podría ser útil como un biomarcador de metilación en este tipo de cáncer 39.
La hipermetilación del ADN como marcador diagnóstico en el cáncer colorrectal
Los mecanismos de hipermetilación descritos anteriormente sugieren que la metilación de determinados genes podría considerarse, en algunos casos, como un evento previo a la aparición del tumor; por tanto, la detección del estado de metilación en el epitelio y en lesiones premalignas, así como en otros tipos de muestras biológicas (sangre o heces), podría utilizarse como un biomarcador de detección temprana para el diagnóstico preventivo y el pronóstico de los pacientes con CCR.
La hipermetilación se ha estudiado como un marcador epigenético en muchos tipos de neoplasias 27,41,42. Por una parte, la aplicación de las técnicas para el análisis de la metilación del ADN para propósitos de diagnóstico podría presentar varias ventajas cuando se compara con otros biomarcadores conocidos, tales como mutaciones puntuales o perfiles de expresión génica. En primer lugar, las alteraciones en los patrones de la metilación del ADN se encuentran en regiones del genoma bien definidas: las islas CpG, y pueden detectarse por diferentes técnicas muy específicas y sensibles; adicionalmente, es importante mencionar que algunas de estas técnicas son económicas 39. En segundo lugar, la posibilidad de realizar el análisis de estos biomarcadores de metilación en sangre periférica y en heces permite evaluar individuos mediante métodos menos invasivos. Por otra parte, la estabilidad del ADN hace posible el análisis de estos biomarcadores en tejidos incluidos en parafina, lo que permite realizar estudios retrospectivos a partir de archivos patológicos de tumores 41.
De acuerdo con lo anterior, la metilación del gen SEPT9 se ha observado en más del 90% de los pacientes con CCR; este es el único biomarcador epigenético aprobado por la Food and Drug Administration (FDA) disponible como una prueba diagnóstica para la detección temprana del CCR 43,44. La detección de SEPT9 metilado en sangre se asocia con un adenocarcinoma colorrectal; en individuos con resultado positivo para esta prueba, se debe recomendar la colonoscopia para confirmar el diagnóstico de CCR. La prueba de metilación de SEPT9 tiene una alta sensibilidad y especificidad, lo que representa una gran ventaja para la detección temprana de CCR 45. Esta prueba se ofrece rutinariamente a los individuos con alto riesgo de CCR en algunos países desarrollados 45. Adicionalmente, también se ha sugerido que el análisis del estado de metilación en muestras de sangre periférica de genes como APC, MLH1, MGMT, CDKN2A y RASSF2A podría ser importante para la detección temprana de CCR 46.
Los biomarcadores epigenéticos descritos, además de tener una importante utilidad en la detección temprana de CCR, podrían ser muy benéficos en el estudio de individuos con un alto riesgo de desarrollar CCR, ya que a estos se les podría recomendar anticipadamente pruebas diagnósticas, intervenciones quirúrgicas o terapias farmacológicas más específicas frente a la naturaleza reversible de las modificaciones epigenéticas 47. Sin embargo, se requieren más estudios que validen las pruebas de los marcadores epigenéticos en el diagnóstico y pronóstico de los pacientes con CCR.
Fenotipo metilador de islas CPG (CIMP) en cáncer colorrectal
El concepto de CIMP (o island methylator phenotype) fue propuesto por Toyota y colaboradores, quienes sugirieron que el CCR se podría clasificar en 2 categorías: una de baja metilación (menos de 2 genes metilados), denominada CIMP-, y otra que exhibe una alta metilación en más de 2 genes simultáneamente o CIMP+48.
Los anteriores autores seleccionaron 5 genes para evaluar el estado CIMP en muestras de CCR: CDKN2A, MINT1, MINT2, MINT31 y MLH1; los cuales son útiles para definir subgrupos de CIMP 49. Además, otros autores han propuesto distintos paneles con un número mayor de marcadores para determinar el estado CIMP como son CACNA1G, CRABP1, IGF2, NEUROG1, RUNX3, SOCS1, HIC1, IGFBP y WR; sin embargo, no existe un consenso sobre cuáles y cuántos genes podría utilizarse para lograr una clasificación adecuada del CIMP en los pacientes con CCR 49-53.
De acuerdo con el análisis de un amplio panel de marcadores de metilación en CCR, se propuso que el CIMP podría dividirse en 2 categorías: CIMP1 y CIMP2 51. Los tumores CIMP1 presentan MSI positiva y se correlaciona con mutaciones en el gen BRAF, mientras que los tumores CIMP2 presentan mutaciones en el gen KRAS y baja frecuencia de mutaciones en TP53, BRAF y MSI negativa 52. Por lo anterior, es de gran importancia resaltar la utilidad de los análisis de metilación en los pacientes con CCR, ya que se tendría la posibilidad de realizar una clasificación molecular más precisa de estos pacientes de acuerdo con sus perfiles de metilación.
Terapia epigenética en cáncer colorrectal
El análisis de la metilación en la región promotora de diversos genes podría proporcionar un marcador pronóstico de la progresión de la enfermedad, así como también de la respuesta a determinados tratamientos antineoplásicos. De esta forma, se ha observado que los marcadores epigenéticos específicos en CCR se asocian con un mal pronóstico. Asimismo, los marcadores epigenéticos han estado en constante investigación debido a la utilidad clínica que tendrían para predecir la respuesta de los pacientes a determinados medicamentos antineoplásicos 10,53.
A diferencia de otras alteraciones moleculares involucradas en el inicio y progresión del CCR como mutaciones y la pérdida alélica, las alteraciones epigenéticas son potencialmente reversibles. Esta característica ha permitido el desarrollo de nuevas terapias basadas en restablecer la actividad de genes silenciados epigenéticamente inhibiendo la metilación del ADN. Actualmente, la 5-azacitidina y 5-azadeoxicitidina son medicamentos inhibidores de la metilación ampliamente investigados y aprobados por la FDA que se utilizan en pacientes con síndrome mielodisplásico, así como también en pacientes con leucemia mielocítica crónica 54,55.
La metilación alterada de la región promotora de diversos genes se plantea como una de las razones por las que los tumores desarrollan resistencia a determinadas terapias antineoplásicas. Una de las ventajas de la terapia epigenética en los tumores colorrectales es que se encuentran hipermetilados varios genes simultáneamente, por lo que este tipo de tratamientos con medicamentos desmetilantes podrían actuar simultáneamente en diversos genes. Adicionalmente, el efecto de la terapia epigenética se podría predecir de manera anticipada, mediante el análisis de genes hipermetilados directamente en el tumor empleando plataformas de secuenciación masiva en paralelo que están disponibles actualmente 54,55.
De acuerdo con lo anteriormente descrito, se ha observado que los pacientes con CCR que presentan el gen MLH1 hipermetilado y, a su vez, la MSI son resistentes a la terapia con 5-fluorouracilo (5-FU) 53; por esta razón, a dichos pacientes se les debe suministrar otro tipo de quimioterapia. De esta manera, se resalta la importancia de determinar el estado de metilación del MLH1 en los pacientes con CCR, porque orienta no solo en la elección de la terapia más adecuada, sino también en el tipo de cirugía que requieren los pacientes.
Adicionalmente, por medio de modelos celulares in vitro, se probó la hipótesis de que con la desmetilación de la región promotora de MLH1 en líneas celulares de CCR se podría restablecer la sensibilidad al tratamiento con 5-FU. Para comprobar esta hipótesis se utilizaron líneas celulares de CCR como SW48 (MLH1 metilado), HCT116 y HCT116 +chr2 (MLH1 mutado), y una línea celular HCT116 +chr3 (MLH1 normal). Se observó que el tratamiento con 5-azacitidina (5-AZA) induce la desmetilación del gen MLH1 y, por consiguiente, el restablecimiento de la expresión del gen y del ARN mensajero (ARNm) en las células SW48. Asimismo, el tratamiento con 5-FU solo reduce el crecimiento celular en la línea HCT116 +chr3, y fue menos efectivo en las demás líneas celulares 56. Estas observaciones corroboran lo establecido en cuanto a que la resistencia al 5-FU puede contrarrestarse mediante el restablecimiento de la expresión del gen MLH1 por el tratamiento con 5-AZA 56. Estos hallazgos tienen un gran impacto para el desarrollo de nuevas terapias epigenéticas antineoplásicas.
Finalmente, el avance de las nuevas tecnologías genómicas masivas permitirá analizar todo el epigenoma con el fin de lograr una mejor caracterización epigenética de los tumores colorrectales, así como también de la utilidad clínica en la búsqueda de nuevos biomarcadores epigenéticos para la detección temprana del CCR.

