










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.33 no.3 Bogotá July/Sept. 2018
https://doi.org/10.22516/25007440.188
Review articles
Cholangiocarcinoma in patients with primary sclerosing cholangitis
1 Estudiante de Medicina, grupo de Gastrohepatología, Universidad de Antioquia, Medellín, Colombia
2 Profesor asociado departamento de Medicina Interna y grupo de Gastrohepatología, Universidad de Antioquia. Hepatólogo, Hospital Pablo Tobón Uribe, Medellín, Colombia
Primary sclerosing cholangitis (PSC) is an uncommon inflammatory disease that affects the bile ducts to produce cholestasis. Currently, the only treatment available is management of symptoms with ursodeoxycholic acid and liver transplantation. In addition to its relation to long-term cirrhosis, its natural history has an important association with cholangiocarcinoma (CCA), an aggressive neoplasia which is almost exclusive to this group of patients. Today CCA is the main cause of death of these patients, primarily due to the difficultly of diagnosis and the very limited treatment options.
This article discusses current ideas about this pathology and emphasizes the importance of appropriate screening with effective diagnostic tests (CA 19-9 and magnetic resonance cholangiopancreatography (MRC)) that help differentiate CCA from benign processes and can detect it in early stages when the probability of curative treatment is much greater.
Keywords: Primary sclerosing cholangitis; cholangiocarcinoma
La colangitis esclerosante primaria (CEP) es una enfermedad inflamatoria poco común que afecta los conductos biliares, produciendo colestasis. Actualmente, el único tratamiento disponible es el manejo sintomático con ácido ursodesoxicólico y el trasplante hepático. Además de ser una etiología de cirrosis a largo plazo, en su historia natural tiene una asociación importante con el colangiocarcinoma (CCA), una neoplasia agresiva que es casi exclusiva de este grupo de pacientes, y hoy en día constituye su principal causa de muerte debido a que es de difícil diagnóstico y cuenta con opciones muy limitadas de tratamiento.
En el presente artículo se exponen conceptos actuales sobre esta patología, enfatizando la importancia de realizar una adecuada tamización con pruebas diagnósticas efectivas (CA 19-9 y colangiorresonancia) que ayuden a diferenciar el CCA de procesos benignos y poder detectarlo en estadios tempranos donde la probabilidad de tratamiento curativo es mucho mayor.
Palabras clave: Colangitis esclerosante primaria; colangiocarcinoma
Introduction
Primary sclerosing cholangitis (PSC), first described in the 1850s, is a chronic, progressive, fibrosing and inflammatory disease that mainly affects the biliary epithelium. It produces stenosis, multifocal dilatations and destruction of the ducts of the entire biliary tree but especially of the medium and long ducts. This leads to cholestasis. 1,2,3,4
Usually, patients are asymptomatic, but clinical manifestations commonly reported at the time of diagnosis include fatigue, fever, jaundice, pruritus and vague upper abdominal discomfort. 5 This pathology’s natural history can vary a great deal, but affected individuals eventually develop complications such as biliary cirrhosis and intra-abdominal malignancies. 1,2,4,5 Currently there is no effective therapy, so the only treatment used is symptomatic management with ursodeoxycholic acid and liver transplantation. Nevertheless, the pathology recurs in 30% to 50% of patients within 10 years of transplantation which is a poor prognosis.2,5
In the pre-transplant era, the main cause of death of PSC patients was liver failure, but currently it is cholangiocarcinoma (CCC). This aggressive and malignant neoplasm originates in cholangiocytes that may have intrahepatic, extrahepatic or hilar locations. The latter is also known as Klatskin’s tumor and is the most common accounting for approximately two thirds of all cases reported. 6,7,8
Patients with PSC have a 5% to 10% lifetime risk of developing CCC: 398 times higher than the risk of the general population: 6,9 While CCC is a rare gastrointestinal malignant tumor for the general population, for patients with PSC this comorbidity is of great importance and is the main cause of death.6,10
Epidemiology
Although epidemiological data for PSC have significant geographical variation, recent studies of the general populations of the Netherlands, the USA, and the United Kingdom have reported prevalences ranging from 3.85 to 6 cases per 100,000 inhabitants and incidences ranging from 0.1 to 0.5 cases per 100,000 inhabitants. This suggests that the geographical variation initially described was skewed because data came from studies carried out in health centers that specialized in liver diseases. 6,11,12
The association of PSC with inflammatory bowel disease (IBD) is well known and occurs in 60% to 80% of cases. It usually appears early and almost always manifests as ulcerative colitis on the right side.5,11 PSC PSC patients are at four times the risk of death than are people in the general population. The main causes of death for these patients are cholangiocarcinoma (CCC) (32%), liver failure (18%), complications of liver transplantation (9%), and colorectal cancer (CRC) (8%). 13
PSC is the most common predisposing condition for CCC in the Western world. 9 CCC occurs in 1% to 2% of PSC patients each year after diagnosis. CCC is often detected within the first three years after the initial diagnosis and has a cumulative risk of 20% thirty years later.5,13 Approximately 50% of CCC cases are diagnosed jointly with PSC or in the year after PSC is diagnosed.13,14 Nevertheless, the incidence of CCC in patients with PSC does not correlate with diagnosis at an early age, since the disease has a less aggressive phenotype in these cases. When PSC is detected later in life, it is more likely to be associated with hepatobiliary malignancies and liver decompensation. 15 Consequently, it is necessary to have a high index of suspicion of CCC once PSC is detected in older people.6,15,16
In addition, it has been documented that the simultaneous presence of IBD, especially ulcerative colitis, increases the risk of cholangiocarcinoma because the symptoms that are accompanied by Crohn’s disease have more benign behavior.6,17 In terms of overall mortality, malignant hepatobiliary neoplasms account for 13% of cancer-related deaths. Of these, 10% to 20% are attributed to the CCC. Mean age of diagnosis is 50 years old, and CCC usually occurs before cirrhosis when it is associated with PSC (Figure 1). 13,18
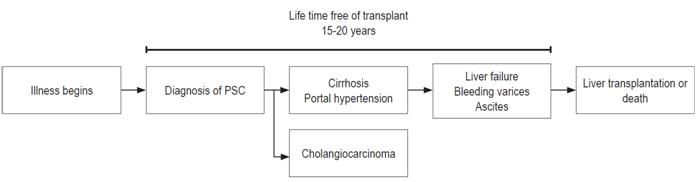
Figure 1 Natural history of primary sclerosing cholangitis. CCC in patients with PSC is often detected within the first one to three years after initial diagnosis at an average age of 50 years. It usually occurs earlier than cirrhosis. PSC: primary sclerosing cholangitis.
PSC’s incidence varies widely in different parts of the world depending on local risk factors. Incidence rangers from 0.5 to 1.5 cases per 100,000 inhabitants in the western world where PSC is the most important risk factor, but is 113 cases per 100,000 in Thailand due to the endemic presence of the Opisthorchis viverrini, a trematode parasite that has been recognized as a causative agent of CCC. 16
Opisthorchis viverrini is a trematode or stave that has been considered to be a human carcinogen by the International Agency for Research on Cancer since a positive direct correlation was found between the incidence of CCC and the prevalence of O. viverrini infections in various regions of Thailand. It infects humans as well as cats and dogs that consume raw or undercooked freshwater fish, especially carp (Cyprinidae family), in their diet (second intermediate host). The metacercariae, the encysted larva, of the parasite contained in the fish flesh is ingested by a person (definitive host) and hatch in the duodenum. They migrate through the ampulla of Vater to the bile duct where they mature and become adults that can live up to 20 years. There they generate asymptomatic chronic infections (20-40 years), advanced periductal fibrosis, cholecystitis and/or CCC 19,20,21.
Eight million people are infected with this parasite, mainly in Thailand, Laos, Cambodia, Vietnam, China and Korea. Five thousand of them have CCC (figures from Thailand). It develops through three key mechanisms: mechanical damage in the epithelium of the ducts caused by alimentary activities of the parasite, the immune response to the infection which leads to inflammation-regeneration-fibrosis, and molecules excreted/secreted by the parasite that cause toxic or carcinogenic effects in the host. One of these substances is granulin, a parasite growth factor that causes human cells to proliferate. 19,20,21
Currently there is no vaccine against infection by this parasite. However, pharmacological treatment with a single 40 mg/kg dose of praziquantel, an anthelmintic used in preventive chemotherapy, has a cure rate of 96%. Nevertheless, excessive use of this medication can reduce its efficacy and induce inflammation of the biliary system. In addition, despite the fact that the parasite is eliminated from the organism, the hepatobiliary changes generated are not always resolved in those treated and reinfections occur frequently. 19,20,21 This is information of great importance since consumption of raw and imported fish in our environment has increased in recent years. 22
Pathogenesis
Although the pathophysiological mechanisms that lead to the development of PSC are not completely known, it is recognized that it is a disease caused by environmental triggers in genetically susceptible people. 23 To date, existence of a genetic predisposition mainly associated with some variants of HLA has been reported. Possibly, a bacterial translocation alters the microenvironment of hepatocytes occurs. This, plus toxicity caused by retained bile and the development of an abnormal immune response are critical factors in the genesis of this pathology. 16,24,25 All of these factors lead to the establishment of a continuously inflamed microenvironment that alters many things including cell division. The sequence of inflammation-dysplasia-carcinoma is probably the origin of CCC in people with PSC. Inflammation through DNA damage, evasion of apoptosis, promotion of cell proliferation and neoangiogenesis all work to create a carcinogenic niche. Recent work has highlighted inflammatory pathways essential roles not only in the development of cancer, but also in invasion and migration of tissues. 18
Another more specific factor reported in the literature is activation of nitric oxide synthase mediated by inflammatory cytokines. This damages DNA and inhibits both enzymes responsible for DNA repair and hepatocyte growth factor which has been postulated as a paracrine mediator of the stroma which regulates tumor invasion and metastasis.18 Cholestasis also activates signaling pathways that increase cell proliferation. In addition, mutations in isocitrate dehydrogenase have been associated with increased levels of p53 and DNA hypermethylation which are probably related to the development of cancer.9,18
Diagnosis
Early diagnosis of CCC is the fundamental pillar of comprehensive management of PSC patients because, as described in this article, its aggressive behavior is the main cause of death in this group of patients. Nevertheless, diagnosis is currently a challenge for the treating medical team because CCC can be easily confused with benign alterations of the bile duct. As a result, it is often diagnosed in late stages after neoplastic invasion has already occurred. This limits therapeutic possibilities even more and casts a shadow over the prognosis. 26
Various studies have reported that factors such as smoking, alcoholism and certain genetic variants increase the risk of suffering from CCC.16,27 However, the clinical usefulness of this is low due to the high incidence of this neoplasm in all patients with PSC. For this reason, routine screening is recommended in all cases regardless of the presence or absence of other risk factors. 16
Currently there is no fully sensitive and specific test for diagnosis of CCC, nor are there guidelines that propose an evidence-based approach to monitoring these patients. 28 However, the literature discusses several proposals for this purpose that combine different modalities of diagnostic tests to increase their diagnostic yield (Figure 2).
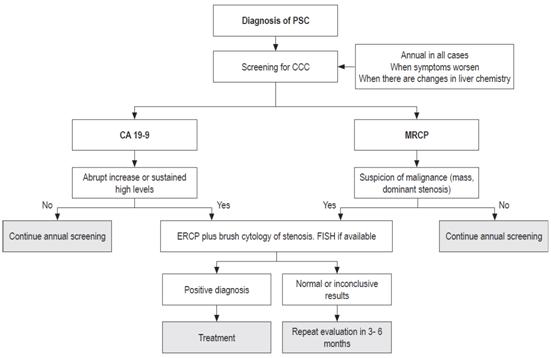
Figure 2 Diagnosis of primary sclerosing cholangitis. PSC: primary sclerosing cholangitis; CCC: cholangiocarcinoma; MRCP: Magnetic resonance cholangiopancreatography; ERCP: endoscopic retrograde cholangiopancreatography; FISH: fluorescent in-situ hybridization.
Monitoring should begin with noninvasive measures such as imaging studies at regular intervals. Hepatobiliary ultrasound or magnetic resonance cholangiopancreatography (MRCP) are useful methods although MRCP is better for identifying small lesions. In addition, it provides information on the intensity and enhancement of lesions which may help differential diagnosis. This is why it is considered the ideal study. 29 In statistical terms, MRCP’s sensitivity is 89%, and its specificity is 75%. Hepatobiliary ultrasound’s sensitivity is 53%, and its specificity is 94% according to a study published in 2008. 30
Either of these strategies can show whether or not there is a dominant mass, polyp, or stenosis of the biliary tree (defined as a narrowing of the diameter of the common bile duct to 1.5 mm or less, and/or narrowing of the diameter of hepatic ducts to 1 mm or less). Any of these findings increase the suspicion of CCC, but do not confirm it. 31 In these cases it is useful to perform endoscopic retrograde cholangiopancreatography (ERCP) accompanied by brush cytology of the stenotic parts to clarify the diagnosis. 32
A metaanalysis covering 747 subjects with PSC that was published in 2014 showed that positive results of brush cytology for CCC had a sensitivity of 43% and specificity of 93%. Nevertheless, it is important to note that this procedure may yield indefinite findings of atypical cells or other suspicious characteristics rather than definitive malignancy. When this happens there are no established guidelines for management. 32
Currently, the most widely used strategy to increase the yield and accuracy of cytology is fluorescence in-situ hybridization (FISH). This tool is based on the ability of nucleic acids to hybridize with complementary strands and works by making probes that bind to specific sequences of the chromosome and emit fluorescence. 33 Among its applications is identification of aneuploidies in the cells of the sample which may suggest the presence of CCC. It was shown that the use of cytology and FISH together for detection of CCC has a sensitivity of 46% and a and specificity of 88% when chromosomal polysomy was found. 26,34
So far, the only biomarker routinely recommended is carbohydrate antigen (CA) 19-9. This serves as an alternative or complement to imaging, but its yield is not the best because in the presence of CCC it may be normal, or in cases of cholangitis it may increase. 26 Various measurements have been proposed as cut-off points for screening tests: values greater than 20 IU/mL report a sensitivity of 78% and a specificity of 67%, while those greater than 130 have a specificity of 100% but a sensitivity of only 13%. 26 However, it should be kept in mind that approximately 7% of patients with PSC present a polymorphism in the FUT3 gene so that they do not express the epitope of CA 19-9. 35
These screening procedures should be performed throughout the course of the disease but especially during the first two years. This is when the incidence of CCC is greatest, when symptoms worsen, and when there are changes in serum cholestatic parameters and rapid or persistent increases in CA 19-9. 26,28
Treatment
Available therapy for CCC can be classified as either curative or palliative. Curative therapy is based on surgical resection, but this is only viable in 25% to 35% of cases since it is only useful for local tumors which have not metastasized to lymph nodes. Liver function should also be preserved since cirrhosis and portal hypertension are markers of a poor prognosis. 36,37
Studies show that 5-year survival rates for resection of well-selected CCC patients range from 27% to 44%. In addition, complementing surgical intervention with adjuvant chemotherapy or radiation therapy to improve the yield of surgery has been postulated. 38
The second curative intervention is liver transplantation although CCC was considered to be an absolute contraindication for transplantation in the past. Now patients with early stages of the disease, especially with perianal CCC of less than 3 cm can receive a transplant. When transplantation is supplemented with adjuvant therapy the rate of survival free of recurrence is 65%. 39,40
There are also local therapeutic strategies such as photodynamic ablation by, external radiation therapy, radioembolization, and high doses of brachytherapy. Nevertheless, most evaluations have been based on small samples of patients or have been retrospective studies that have not allowed for high quality information that could support their use and provides information on possible short and long term adverse effects. 38
Palliative therapy seeks to improve patients’ general conditions and/or lengthen life expectancy. A clinical trial in 2010 showed that systemic chemotherapy, consisting of the combination of cisplatin and gemcitabine, increase life expectancy by three to six months. These results were later reproduced in a Japanese study.41,42 For patients with renal failure, it has been proposed that oxaliplatin replace cisplatin in this scheme. 43 Similarly, gemcitabine monotherapy is recommended in elderly patients whose general health is compromised. A reviewing of the use of second-line drugs has found a proposal to use fluoropyrimidines, but currently there is not enough information to validate this application. 44
Finally, malignant obstruction of the bile duct is a late complication of different types of neoplasms including CCC. Nowadays, endoscopic stenting is recommended for temporary improvement of jaundice and pruritus secondary to obstructions of the bile duct. 26
Perspectives
Bearing in mind that the success of curative therapy depends on the stage at which CCC is diagnosed, current research focuses on the development of efficient and minimally invasive diagnostic methods. These include proteomic studies of urine, serum and bile that detect specific peptide panels correlated with CCC, use of non-coding microRNAs as biliary biomarkers of malignancy, and analysis of extracellular microvesicles in bile samples to differentiate benign from malignant stenoses. 10,45
It has been found that treatment using molecules such as epithelial growth factor and vascular endothelial growth factor has not provided positive results. 44) Consequently, the need to conduct studies that offer better possibilities for patients with PSC and CCC is recognized, and it is hoped that there will be further progress in this or other areas of research.
Referencias
1. Sarkar S. Primary sclerosing cholangitis multiple phenotypes, multiple approaches. Clin Liver Dis. 2016;20(1):67-77. doi: https://doi.org/10.1016/j.cld.2015.08.005. [ Links ]
2. Horsley-Silva JL, Carey EJ, Lindor KD. Advances in primary sclerosing cholangitis. Lancet Gastroenterol Hepatol. 2016;1(1):68-77. doi: https://doi.org/10.1016/S2468-1253(16)30010-3. [ Links ]
3. Eaton JE, Talwalkar JA, Lazaridis KN, Gores GJ, Lindor KD. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology. 2013;145(3):521-36. doi: https://doi.org/10.1053/j.gastro.2013.06.052. [ Links ]
4. Takakura WR, Tabibian JH, Bowlus CL. The evolution of natural history of primary sclerosing cholangitis. Curr Opin Gastroenterol. 2016;33(2):1. doi: https://doi.org/10.1097/MOG.0000000000000333. [ Links ]
5. Gidwaney NG, Pawa S, Das KM. Pathogenesis and clinical spectrum of primary sclerosing cholangitis. World J Gastroenterol. 2017;23(14):2459. doi: 10.3748/wjg.v23.i14.2459. [ Links ]
6. Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, Spanier BWM, Poen AC, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013;58(6):2045-55. doi: https://doi.org/10.1002/hep.26565. [ Links ]
7. Fevery J, Verslype C. An update on cholangiocarcinoma associated with primary sclerosing cholangitis. Curr Opin Gastroenterol. 2010;26(3):236-45. doi: https://doi.org/10.1097/MOG.0b013e328337b311. [ Links ]
8. Plentz RR, Malek NP. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Best Pract Res Clin Gastroenterol. 2015;29:245-52. [ Links ]
9. Rizvi S, Gores GJ. Molecular pathogenesis of cholangiocarcinoma. Dig Dis. 2014;32(5):564-9. doi: https://doi.org/10.1159/000360502. [ Links ]
10. Ehlken H, Zenouzi R, Schramm C. Risk of cholangiocarcinoma in patients with primary sclerosing cholangitis. Curr Opin Gastroenterol. 2017;33(2):1. doi: https://doi.org/10.1097/MOG.0000000000000335. [ Links ]
11. Toy E, Balasubramanian S, Selmi C, Li CS, Bowlus CL. The prevalence, incidence and natural history of primary sclerosing cholangitis in an ethnically diverse population. BMC Gastroenterol. 2011;11(1):83. doi: https://doi.org/10.1186/1471-230X-11-83. [ Links ]
12. Card TR, Solaymani-Dodaran M, West J. Incidence and mortality of primary sclerosing cholangitis in the UK: a population-based cohort study. J Hepatol. 2008;48(6):939-44. doi: https://doi.org/10.1016/j.jhep.2008.02.017. [ Links ]
13. Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Lööf L, Danielsson A, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002;36(3):321-7. doi: https://doi.org/10.1016/S0168-8278(01)00288-4. [ Links ]
14. Boberg KM, Bergquist A, Mitchell S, Pares A, Rosina F, Broomé U, et al. Cholangiocarcinoma in primary sclerosing cholangitis: risk factors and clinical presentation. Scand J Gastroenterol. 2002;37(10):1205-11. doi: https://doi.org/10.1080/003655202760373434. [ Links ]
15. Eaton JE, McCauley BM, Atkinson EJ, Juran BD, Schlicht EM, de Andrade M, et al. Variations in primary sclerosing cholangitis across the age spectrum. J Gastroenterol Hepatol. 2017;32(10):1763-8. doi: 10.1111/jgh.13774. [ Links ]
16. Bergquist A, von Seth E. Epidemiology of cholangiocarcinoma. Best Pract Res Clin Gastroenterol. 2015;29(2):221-32. doi: https://doi.org/10.1016/j.bpg.2015.02.003. [ Links ]
17. Zheng H, Jiang X. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: a meta-analysis of 16 observational studies. Eur J Gastroenterol Hepat. 2016;28:383-90. doi: 10.1097/MEG.0000000000000576. [ Links ]
18. Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145(6):1215-29. doi: https://doi.org/10.1053/j.gastro.2013.10.013. [ Links ]
19. Sripa B, Brindley PJ, Mulvenna J, Laha T, Smout MJ, Mairiang E, et al. The tumorigenic liver fluke Opisthorchis viverrini - Multiple pathways to cancer. Trends Parasitol. 2012;28(10):395-407. doi: https://doi.org/10.1016/j.pt.2012.07.006. [ Links ]
20. Sripa B, Bethony JM, Sithithaworn P, Kaewkes S, Mairiang E, Loukas A, et al. Opisthorchiasis and Opisthorchis-associated cholangiocarcinoma in Thailand and Laos. Acta Trop. 2011;120:S158-68. doi: https://doi.org/10.1016/j.actatropica.2010.07.006. [ Links ]
21. Young ND, Nagarajan N, Lin SJ, Korhonen PK, Jex AR, Hall RS, et al. The Opisthorchis viverrini genome provides insights into life in the bile duct. Nat Commun. 2014;5:4378. doi: https://doi.org/10.1038/ncomms5378. [ Links ]
22. Autoridad Nacional de Acuicultura y Pesca. El consumo de pescado en el país va en aumento. 2017. [ Links ]
23. Pollheimer MJ, Halilbasic E, Fickert P, Trauner E. Pathogenesis of primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2017;25(6):727-39. doi: https://doi.org/10.1016/j.bpg.2011.10.009. [ Links ]
24. Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al. Europe PMC funders group dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. 2013;1(6):670-5. doi: https://doi.org/10.1038/ng.2616. [ Links ]
25. Williamson K, Chapman R. New therapeutic strategies for primary sclerosing cholangitis. Semin Liver Dis. 2016;36(1):005-14. doi: https://doi.org/10.1055/s-0035-1571274. [ Links ]
26. Thosani N. Endoscopic retrograde cholangiopancreatography for primary sclerosing cholangitis cholangioscopy choledochoscopy biliary drainage bile duct sampling. Clin Liver Dis. 2014;18(4):899-911. doi: https://doi.org/10.1016/j.cld.2014.07.013. [ Links ]
27. Boberg KM, Lind GE. Primary sclerosing cholangitis and malignancy. Best Pract Res Clin Gastroenterol. 2011;25(6):753-64. doi: 10.1016/j.bpg.2011.10.002.28. [ Links ]
28. Folseraas T, Boberg KM. Cancer risk and surveillance in primary sclerosing cholangitis. Clin Liver Dis. 2016;20(1):79-98. doi: https://doi.org/10.1016/j.cld.2015.08.014. [ Links ]
29. Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology. 2011;54(5):1842-52. doi: https://doi.org/10.1002/hep.24570. [ Links ]
30. Charatcharoenwitthaya P, Enders FB, Halling KC, Lindor KD. Utility of serum tumor markers, imaging, and biliary cytology for detecting cholangiocarcinoma in primary sclerosing cholangitis. Hepatology. 2008;48(4):1106-17. doi: https://doi.org/10.1002/hep.22441. [ Links ]
31. Stiehl A, Rudolph G, Klöters-Plachky P, Sauer P, Walker S. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002;36(2):151-6. doi: https://doi.org/10.1016/S0168-8278(01)00251-3. [ Links ]
32. Trikudanathan G, Navaneethan U, Njei B, Vargo JJ, Parsi MA. Diagnostic yield of bile duct brushings for cholangiocarcinoma in primary sclerosing cholangitis: a systematic review and meta-analysis. Gastrointest Endosc. 2014;79(5):783-9. doi: https://doi.org/10.1016/j.gie.2013.09.015. [ Links ]
33. Rodríguez Martínez R, Suescún Otero G. Aplicaciones e Inconvenientes de la técnica hibridación in situ fluorescente (FISH) en la identificación de microorganismos. Salud Uninorte. 2013;29(2):327-340. [ Links ]
34. Barr Fritcher EG, Kipp BR, Voss JS, Clayton AC, Lindor KD, Halling KC, et al. Primary sclerosing cholangitis patients with serial polysomy fluorescence in situ hybridization results are at increased risk of cholangiocarcinoma. Am J Gastroenterol. 2011;106(11):2023-8. doi: https://doi.org/10.1038/ajg.2011.272. [ Links ]
35. Wannhoff A, Hov JR, Folseraas T, Rupp C, Friedrich K, Anmarkrud JA, et al. FUT2 and FUT3 genotype determines CA19-9 cut-off values for detection of cholangiocarcinoma in patients with primary sclerosing cholangitis. J Hepatol. 2017;59(6):1278-84. doi: https://doi.org/10.1016/j.jhep.2013.08.005. [ Links ]
36. Neumann UP, Schmeding M. Role of surgery in cholangiocarcinoma: from resection to transplantation. Best Pr Res Cl Ga. 2015;29(2):295-308. doi: https://doi.org/10.1016/j.bpg.2015.02.007. [ Links ]
37. Ajani JA, Correa AM, Walsh GL, Komaki R, Lee JH, Vaporciyan AA, et al. Trimodality therapy without a platinum compound for localized carcinoma of the esophagus and gastroesophageal junction. Cancer. 2010;116(7):1656-63. doi: https://doi.org/10.1002/cncr.24935. [ Links ]
38. Vogel A, Dudeck O. Is there any evidence for a role of local treatment in cholangiocarcinoma? Viszeralmedizin. 2014;30(4):254-60. doi: https://doi.org/10.1159/000365312. [ Links ]
39. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51(2):660-78. doi: 10.1002/hep.23294. [ Links ]
40. Darwish Murad S, Kim WR, Harnois DM, Douglas DD, Burton J, Kulik LM, et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology. 2012;143(1):88-98.e3. doi: https://doi.org/10.1053/j.gastro.2012.04.008. [ Links ]
41. Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. New Engl J Med. 2010;362(14):1273-81. doi: https://doi.org/10.1056/NEJMoa0908721. [ Links ]
42. Okusaka T, Nakachi K, Fukutomi A, Mizuno N, Ohkawa S, Funakoshi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Brit J cancer. 2010;103(4):469-74. doi: https://doi.org/10.1038/sj.bjc.6605779. [ Links ]
43. André T, Reyes-Vidal JM, Fartoux L, Ross P, Leslie M, Rosmorduc O, et al. Gemcitabine and oxaliplatin in advanced biliary tract carcinoma: a phase II study. Brit J Cancer. 2008;99(6):862-7. doi: https://doi.org/10.1038/sj.bjc.6604628. [ Links ]
44. Vogel A, Saborowski A. Cholangiocellular carcinoma. Digestion. 2017;95(3):181-5. doi: https://doi.org/10.1159/000454763. [ Links ]
45. Berretta M, Cavaliere C, Alessandrini L, Stanzione B, Facchini G, Balestreri L, et al. Serum and tissue markers in hepatocellular carcinoma and cholangiocarcinoma: clinical and prognostic implications. Oncotarget. 2017;8(8):14192-220. doi: https://doi.org/10.18632/oncotarget.13929. [ Links ]
Received: December 11, 2017; Accepted: February 22, 2018
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
