











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La enfermedad de Ménétrier (EM) se describió por primera vez en 1888 por el patólogo francés Pierre Eugene Ménétrier en pacientes adultos. La presentación de la enfermedad en la población infantil, es rara, con menos de 100 casos publicados en la literatura 1. Se caracteriza por un engrosamiento de pliegues gástricos con hipoalbuminemia secundaria a la pérdida de proteínas a través de la mucosa gastrointestinal. La mayoría de los casos pediátricos se asocian a infecciones virales y dentro de ellas es el citomegalovirus (CMV) el agente etiológico más mencionado. A diferencia de la enfermedad del adulto, es un trastorno agudo con curso benigno y autolimitado.
Se debe sospechar esta enfermedad en pacientes con síntomas gastrointestinales inespecíficos, hipoproteinemia y edemas, en los que se hayan excluido causas hepáticas, renales y cardíacas 2.
Caso clínico 1
El primer paciente fue un varón de 23 meses con antecedentes personales de asma y rinitis alérgica, que fue remitido a nuestro hospital para un estudio de hipoalbuminemia. Dos semanas antes comenzó con cuadro gripal y fiebre, luego asociado con vómito, distensión abdominal y diarrea. Consultó a otros centros asistenciales donde realizaron tratamiento sintomático. Al décimo día de evolución presentó deterioro clínico dado por astenia, adinamia, hiporexia y edema generalizado con ganancia de peso objetiva de 4 kilos.
Al ingreso se observaba en regulares condiciones generales, sus signos vitales eran tensión arterial (TA): 95/41/59, frecuencia cardíaca (FC): 140 latidos por minuto (lpm), frecuencia respiratoria (FR): 24 respiraciones por minuto (rpm), saturación de oxígeno (SatO2): 98%, peso: 15 kg y talla: 92 cm. Lucia pálido, con signos de deshidratación leve, polipneico y con rinorrea hialina. Era llamativo el edema generalizado. Fue hospitalizado en unidad de cuidados intermedios con impresión diagnóstica de síndrome nefrótico.
Las ayudas diagnósticas iniciales fueron hemograma con serie roja y plaquetaria normales, y serie blanca con 18300 leucocitos con 27% de linfocitos reactivos, velocidad de sedimentación globular (VSG): 2 y proteína C-reactiva (PCR): 0,83. El nivel de triglicéridos fue de 220, el estudio de coagulación fue normal, la función renal fue normal (creatinina: 0,83 mg/dL), función hepática normal (alanina-aminotransferasa [ALT]: 10 UI/L, aspartato-aminotransferasa [AST]: 31 UI/L), con hipoproteinemia (proteínas totales: 3,1 g/dL) y con hipoalbuminemia (1,9 g/dL). El estudio de orina fue normal, sin evidencia de proteinuria ni hematuria.
La serología a hepatitis B (VHB), hepatitis C (VHC), virus de Epstein-Barr (VEB), virus de la inmunodeficiencia humana (VIH), leptospira, micoplasma, dengue y toxoplasma fueron negativas; y resultó positiva tanto para inmunoglobulina M (IgM) como inmunoglobulina G (IgG) para citomegalovirus (CMV).
Se realizó una ecografía de abdomen que reportó derrame pleural bilateral, líquido libre en el espacio perirrenal y periesplácnico.
El paciente fue evaluado por nefrología infantil, que descartó causas renales que explicaran los edemas; igualmente por hepatología, que descartó una trombosis portal aguda que pudiera estar explicando el cuadro. Por el hallazgo de infección por CMV se interconsultó también a infectología, que consideró que a pesar de la evidencia de infección (carga viral de CMV: 9932 copias/mL), el paciente no ameritaba tratamiento antiviral ya que no se demostró compromiso de órgano blanco y era mayor el riesgo de toxicidad por el medicamento.
Finalmente, se interconsultó a gastropediatría al sexto día de hospitalización y se ordenó una endoscopia digestiva superior (EDS) y colonoscopia por la sospecha de gastropatía y/o enteropatía perdedora de proteína. Se encontraron pliegues corporales hipertróficos, erosionados y con mucosa friable, mucosa antral de aspecto normal y mucosa fúndica con erosiones superficiales, por lo que se sospechó EM (Figura 1). Con estos hallazgos se mantiene una conducta expectante con respecto a la realización de colonoscopia.
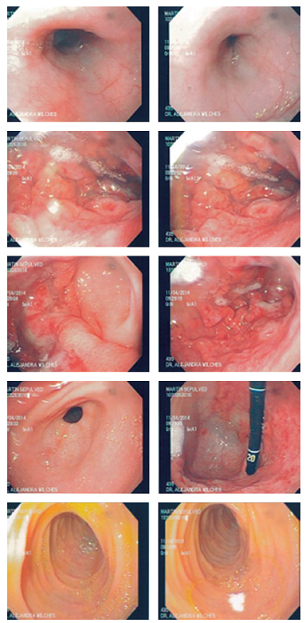
Figura 1 Mucosa esofágica y duodenal de aspecto normal. Pliegues corporales hipertróficos y erosionados.
En el estudio anatomopatológico se encontró esofagitis leve, gastropatia hipertrófica (hiperplasia foveolar, dilatación y atrofia de glándulas, aumento de fibras musculares en la lámina propia, sin Helicobacter pylori) y signos de duodenitis crónica, hallazgos que confirmaron el diagnóstico de EM (Figura 2).
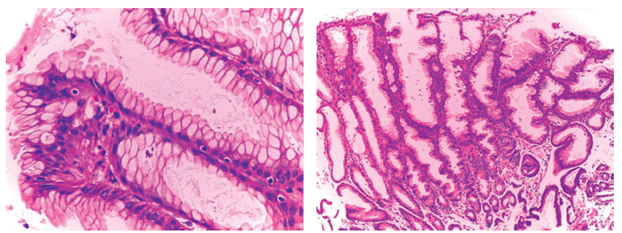
Figura 2 Elongación e hiperplasia de fovéolas, aumento de fibras musculares en lámina propia e infiltrado inflamatorio de predominio mononuclear (200x y 100x).
La evolución del paciente fue favorable. Inicialmente, se trató con infusiones de albúmina y diurético y se logró la mejoría de los edemas. Recibió también tratamiento antisecretor con esomeprazol y soporte nutricional. Fue dado de alta al día 17 de hospitalización y volvió a revisión por consulta externa a los 15 días, asintomático y con valor de albúmina sérica de 3,6 mg/dL. Se programó para EDS de control que no se realizó por decisión de sus padres, pero continuó asistiendo a revisiones periódicas sin reporte clínico de recaída.
Caso clínico 2
El segundo paciente fue un varón de 5 años de edad quien consultó por cuadro clínico de 15 días de evolución de dolor abdominal asociado con múltiples episodios de vómito y 3-4 deposiciones diarreicas por día, con moco, sin sangre, por lo general posprandiales. Posteriormente, presentó edema bipalpebral, oliguria y orina espumosa. Fue hospitalizado en una institución de segundo nivel para estudio, donde descartaron síndrome nefrótico y causas hepáticas que explicaran los edemas. Realizaron TAC de abdomen en el que encontraron múltiples adenopatías mesentéricas y engrosamiento de las asas del yeyuno e íleon, por lo que lo remitieron a un centro de tercer nivel para estudios y, por ese motivo, ingresó a nuestra institución.
Los signos vitales al ingreso fueron: FC: 130 lpm, FR: 24 rpm, SatO2: 97%, TA: 69/45/56, peso: 19,7 kg y talla: 112 cm. Tenía edemas generalizados, con crépitos basales derechos, con onda ascítica positiva y circulación colateral en hemiabdomen superior.
Se realizaron paraclínicos: hemograma con serie roja y plaquetaria normal, leucocitos: 15 100, en extendido de sangre periférica se observó 2% de linfocitos reactivos, uroanálisis normal, función renal normal (creatinina: 0,53 mg/dL, nitrógeno ureico sanguíneo [BUN] 18 mg/dL), función hepática y pruebas de coagulación normales, triglicéridos: 135 mg/dL e hipoproteinemia (proteínas totales séricas: 2,7 g/dL y albúmina 1,6 g/dL). Igualmente, se realizó una radiografía de tórax con derrame pleural derecho en moderada cantidad y ecografía de abdomen sin alteraciones.
Fue evaluado por un gastropediatra, quien sospechó gastropatía perdedora de proteínas frente a enfermedad inflamatoria intestinal (por los hallazgos en TAC de abdomen), por lo que se programó para EDS y colonoscopia. Se encontraron pliegues corporales muy engrosados, mucosa corpoantral friable con pseudopólipos y erosiones, mucosa antral de aspecto folicular, bulbo duodenal y segunda porción con mucosa de aspecto normal, y la mucosa colónica tenía aspecto normal en toda su extensión (Figura 3).
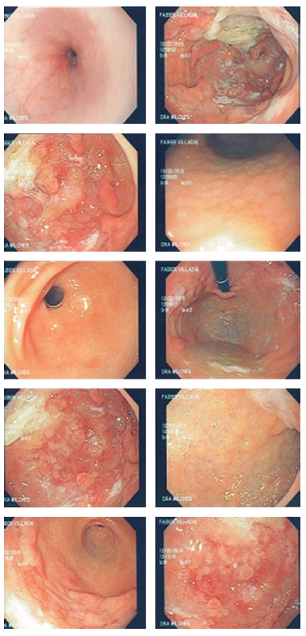
Figura 3 Pliegues gástricos hipertróficos, con pseudopólipos y friabilidad, y mucosa antral folicular. Secreción gástrica viscosa y espesa.
Con estos hallazgos se sospechó EM y se solicitaron anticuerpos para CMV, los cuales reportaron con IgG e IgM reactivos, pero con carga viral para CMV negativa (77 ácido desoxirribonucleico [ADN]/mL).
El paciente evolucionó favorablemente luego del tratamiento diurético, la reposición con albúmina endovenosa, antisecretor y manejo nutricional; después de 9 días de hospitalización, se dio el alta para el manejo ambulatorio con esomeprazol y recomendaciones nutricionales. Asistió a consulta un mes después completamente asintomático y se revisaron los resultados de biopsia que reportó una gastropatía hipertrófica compatible con EM, 60 eosinófilos por campo y H. pylori negativo (Figura 4). Biopsias de mucosa colónica: inflamación crónica inespecífica.
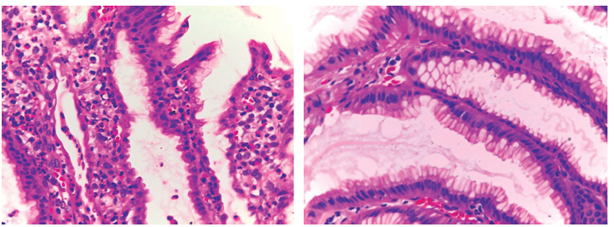
Figura 4 Arquitectura alterada por marcada elongación y dilatación de fovéolas, revestidas por el epitelio cilíndrico hiperplásico con infiltrado inflamatorio mono y polimorfonucleares, neutrófilos y eosinófilos (100x y 200x).
Se indicó continuar el tratamiento antisecretor y se solicitó EDS de control que no se realizó ya que no volvió al seguimiento.
Discusión
La EM es un trastorno gástrico adquirido que se caracteriza por pliegues hiperplásicos gigantes, exceso de secreción de moco e hipoproteinemia debido a la pérdida selectiva de las proteínas del suero a través de la mucosa gástrica. El curso clínico de la enfermedad en los adultos es de inicio insidioso y progresivo, y se asocia con malignidad; pero en los niños, el trastorno se caracteriza por la aparición brusca y resolución espontánea de 2 a 10 semanas solo con tratamiento de soporte 3.
Su causa es desconocida, pero la literatura ha descrito una posible asociación con irritantes químicos, toxinas, factores dietéticos, factores neuroemocionales, endocrinológicos, procesos alérgicos o con alteraciones inmunológicas.
La asociación con procesos infecciosos se ha relacionado fuertemente con CMV (hasta en el 70% de los casos) 4,5,6, aunque también hay descripciones asociadas con H. pylori7, Herpes simple, Giardia intestinalis y Mycoplasma pneumoniae, entre otros 3.
Las investigaciones recientes sugieren que la fisiopatología puede estar explicada por el estímulo que producen los desencadenantes sobre el receptor del factor de crecimiento epidérmico (EGFR), que es un receptor transmembrana con actividad de tirosina-cinasa, el cual a su vez estimula la vía de señalización del factor de crecimiento transformante alfa (TGF-α), que finalmente va a generar el sobrecrecimiento de las células epiteliales mucosas de predominio en cuerpo y fondo gástrico 6,7,8.
La EM infantil afecta habitualmente a menores de 6 años y suele presentar un comienzo agudo. Los síntomas iniciales son inespecíficos, similares a cualquier cuadro viral, con fatiga, náuseas, vómitos y dolor abdominal. Cuando se presenta la hipoalbuminemia, se produce edema periférico y posteriormente ascitis, derrame pleural e incluso pericárdico; los pacientes suelen presentarse al servicio de urgencias en anasarca, como fue evidente en los 2 casos mencionados 6. Analíticamente, es característica la presencia de hipoproteinemia e hipoalbuminemia, lo que obliga a descartar compromiso renal y hepático 9.
El diagnóstico de la EM se realiza por un estudio endoscópico en el cual se observan pliegues engrosados en el fondo y cuerpo gástrico, mucosa gástrica ligeramente mamelonada o nodular y con áreas pequeñas de tejido ulcerado o necrótico; además, se puede ver un incremento de la secreción gástrica de característica espesa, viscosa y transparente 10. Durante la EDS, es necesario tomar muestras para hacer correlación con el estudio histopatológico. Los hallazgos son similares a la EM del adulto, caracterizándose por hipertrofia de pliegues gástricos e hiperplasia foveolar, con engrosamiento de la mucosa por proliferación, elongación y dilatación quística de glándulas gástricas; y con hipersecreción, edema e infiltrado inflamatorio 11,12.
La asociación con infección aguda por CMV es compleja. La evidencia de relación con la infección por CMV se establece por el hallazgo de células infectadas en la mucosa gástrica mediante técnicas de PCR, detección de antígenos virales por inmunohistoquímica, cultivo viral de mucosa gástrica o la visualización de inclusiones virales típicas. La detección de antigenemia, PCR en sangre o las serologías son menos sensibles en la detección de la afectación gastrointestinal; sin embargo, permiten detectar la infección y establecer la sospecha diagnóstica. La hibridación in situ y la inmunohistoquímica se han propuesto como las técnicas de laboratorio más sensibles, pero existen casos en los que no se ha identificado la presencia del virus. El problema es que, en estos casos, la infección por CMV puede corresponder a una primoinfección, reactivación o reinfección. La excreción viral en la orina y la serología de IgM positiva pueden mantenerse durante meses, por lo que su hallazgo aislado no permite confirmar que la infección por CMV sea la causa de la EM 2.
Por el curso benigno y autolimitado de la EM en los niños, se determina que solo se hace tratamiento antiviral con ganciclovir a aquellos pacientes en quienes se logre demostrar la presencia del CMV en la mucosa gástrica y no tengan mejoría clínica y paraclínica con las medidas de soporte que incluyen reposición hídrica, infusión de albúmina y diuréticos, asociados con antisecretores y medidas dietéticas 13. Experimentalmente, en adultos se han hecho ensayos con anticuerpos monoclonales contra el EGFR en casos refractarios; incluso la literatura menciona un caso refractario y atípico en niños en el cual usaron octreótido con éxito 14.
Otras causas de gastropatías y enteropatías perdedoras de proteínas a tener en cuenta en el diagnóstico diferencial son la enfermedad celíaca, el linfoma gástrico, la gastroenteritis eosinofílica, la gastropatía hipertrófica asociada con H. pylori y la enfermedad de Crohn.
La gastroenteritis eosinofílica puede incluir antro gástrico e intestino delgado, pero a menudo hay antecedentes familiares de enfermedad alérgica. El linfoma gástrico es muy raro en niños y no está relacionado con edema e hipoproteinemia. La enfermedad de Crohn y el síndrome de Peutz-Jeghers tienen diferentes signos y síntomas. Otras enfermedades raras que tienen signos radiológicos similares con EM son síndrome de Zollinger-Ellison, gastritis secretora hipertrófica, varices gástricas y linfangiectasia intestinal 15.
Conclusiones
Presentamos 2 casos clínicos típicos de EM infantil que llegaron a nuestro centro asistencial con comportamiento y curso clínico similar al que describe la literatura mundial y podemos concluir que se debe sospechar una EM en caso de que un niño tenga edemas e hipoalbuminemia sin compromiso renal, sin disfunción hepática ni cardíaca, asociado con hipertrofia de pliegues gástricos e hiperplasia foveolar.

