










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.34 no.1 Bogotá Jan./Mar. 2019
https://doi.org/10.22516/25007440.225
Original articles
Relationship of Methylation of Tumor Suppressor Genes with Clinical Stage of Colorectal Cancer in Venezuelan Patients
1Universidad de Zulia, Instituto de Investigaciones Genéticas. Maracaibo, Venezuela
2Universidad de Zulia, Departamento de morfofisiopatología. Escuela de Bioanálisis. Maracaibo, Venezuela
3Universidad de Zulia, Instituto de Investigaciones Genéticas. Laboratorio de genética molecular. Maracaibo, Venezuela
4Universidad de Zulia, Facultad de Medicina, Escuela de Bioanálisis. Cátedra práctica profesional de hematología. Maracaibo, Venezuela
5Universidad de Zulia, Facultad de Medicina, Instituto de Investigaciones Genéticas. Laboratorio de citogenética y genética de cáncer. Maracaibo, Venezuela
Colorectal cancer is a heterogeneous disease which involves hereditary and environmental factors. The inherited forms have genes which are responsible for increasing the tumor development in carriers. Environmental factors are considered responsible for many sporadic forms. The objective of this study was to analyze the methylation status of five genes involved in colorectal carcinogenesis and their relationships with the various clinical stages of these tumors. Our analysis revealed that the methylation status of the promoters of genes HMLH1, APC, P15, P16 and CDH1, considered to be among the earliest alterations in this process, ranged from 13.3% for HMLH1 to 56.6% for APC. In addition, epigenetic inactivation of APC and P16 genes could be responsible for the appearance and progression of tumors since inactivation was found in stage II patients. On the other hand, the APC and p15 gene were mutated in all stages of carcinogenesis, so they could be involved throughout the processes of initiation, invasion and metastasis. Finally, our results support using identification of methylation of suppressor genes since they identify epigenetic targets for development of new chemotherapy treatments. This is emerging as a strategy with great potential since epigenetic alterations are, in principle, potentially reversible.
Keywords: Epigenetics; colorectal cancer; clinical evolution
El cáncer colorrectal es una enfermedad heterogénea, en cuya aparición se involucran factores hereditarios y ambientales. En las formas heredadas existen genes responsables de incrementar el desarrollo tumoral en los portadores, y se consideran a los factores medioambientales como responsables de gran parte de las formas esporádicas. El objetivo de este estudio fue analizar el estado de metilación de 5 genes implicados en la carcinogénesis colorrectal y su relación con los distintos estadios clínicos de estos tumores. Por una parte, nuestro análisis reveló que el estado de metilación de los promotores de los genes HMLH1 (human mut homologue 1), APC (adenomatous poliposis coli), P15, P16 y CDH1, considerados como unas de las alteraciones más tempranas en este proceso; fluctuaron entre 13,3 % para hMLH1 y 56,6 % para APC. También reveló que la inactivación epigenética de los genes APC y P16 podrían ser responsables de la aparición y de la progresión de los tumores ya que se encontraron en pacientes con estadio II. Por otra parte, los genes APC y p15 resultaron estar mutados en todas las etapas de la carcinogénesis, por lo que se involucrarían en todos los procesos tanto de inicio como de invasión y metástasis. Por último, nuestros resultados apoyan la utilización de la identificación de la metilación de los genes supresores ya que se están identificando dianas epigenéticas para el desarrollo de nuevos tratamientos de quimioterapia y está emergiendo como una estrategia con gran potencial dado que, en principio, las alteraciones epigenéticas son potencialmente reversibles.
Palabras clave: Epigenética; cáncer colorrectal; evolución clínica
Introduction
Colorectal cancer (CRC) consists of uncontrolled growth of the mucosal cells of these intestinal regions. It is considered one of the biggest public health problems in many developed countries and is the second leading cause of cancer in the United States. This seems controversial since it is considered to be curable when diagnosed early. In 2018, the EFE news service in Bogotá reported that this type of cancer causes approximately 49,000 cases a year and that he highest rates in Latin America are found in Uruguay (29.5 new cases per year per 100 000 inhabitants), Argentina (23.8/100 000), Cuba (19.7/100 000), Costa Rica (16.4/100 000), Brazil (15.8/100 000) and Chile (15/100 000) while Guatemala (4.3/100 000) and Honduras (6.9/100 000) report the lowest rates. 1 Countries in the intermediate range include Colombia (12.9/100 000), Panama (12.5/100 000), Paraguay (12.1/100 000), Peru (11.1/100 000), Ecuador (10, 7/100 000), Dominican Republic (10.2/100 000), Bolivia (9.1/100 000), Nicaragua (7.9/100 000) and Mexico (7.8/100 000). According to the mortality statistics of the Venezuelan Ministry of Popular Power for Health (MPPS), in 2012 there were 1,474 cases in men with 756 deaths and 1,661 cases in women with 801 deaths. 2 According to statistics from the Venezuelan Oncology Society, in 2015 around 3,500 cases were diagnosed throughout the country. 3
CRC is characterized by genetic and epigenetic deregulation of signals that occur in multi-step transduction cascades. Long-term accumulation of these changes produces epigenetic alterations that lead to the appearance of CRC 4.
Clinical-pathological staging is the best way to predict the course of the disease, but this system offers only gross estimates that lead to unnecessary treatment of a large number of patients while recurrences occur among patients who only received surgery. 5 The risk of CRC increases with age, so given that the world’s population is growing and aging, the coming decades will put unprecedented pressure on the health care institutions around the world. For these reasons, it is necessary to identify molecular biomarkers to guide clinical decision making on how to stratify patients for optimal treatment regimens. In particular, this is needed for patients with stage II CRC. They are not usually offered adjuvant therapy even though about 20% to 30% relapse and die within five years of surgery. Similarly, stage III patients over the age of 75 do not routinely receive adjuvant therapy even though the evidence suggests that there is a benefit from such treatment. 6
Prognostic biomarkers that distinguish between high risk and low risk patients in these stages are highly justified. Epigenetic regulation of gene function plays an important role in development, and methylation is the mechanism that ensures the specific expression of cell types of a limited group or spectrum of genes. 7 Various diseases, including cancer, present altered patterns of methylation that change the perfect epigenetic balance that exists in normal cells. 8 Methylation of deoxyribonucleic acid (DNA) is an efficiently regulated dynamic process in which unmethylated sequences can be methylated and methyl groups can be lost. Methylation patterns of somatic cells are the results of both methylation and demethylation. The DNA methylation reaction is catalyzed by DNA methyltransferases and involves the transfer of the methyl group of S-adenosyl-L-methionine to the carbon 5 of the cytosine. 9 Three different enzymes that carry out this reaction have been identified. Known as DNA methyltransferases (DNMTs), DNMT 1 is responsible for maintenance DNA methylation while DNMT3A and DNMT3B are involved in de novo methylation. In humans, de novo activity adds a methyl group to the cytosine of the unmethylated CpG dinucleotide to create a new, highly hemimethylated, CpG. 10
This aberrant pattern of methylation is characterized by overall hypomethylation of the genomic DNA but hypermethylation of the promoter areas. The former leads to genomic instability while the latter leads to gene silencing. 11,12 The transcriptional repression associated with aberrant methylation of the CpG islets in promoter areas is the consequence of intense changes in the structure of chromatin produced by the interaction of 5-methylcytosine with different protein complexes that recruit histone-modifying enzymes such as histone acetyltransferase and histone methyltransferase. 12
Genes involved in many metabolic pathways can undergo aberrant methylation. These genes include DNA repair genes (human mut homologue 1, hMLH1), cell cycle regulators such as APC (adenomatous polyposis coli), APC and DCC (deleted in colorectal cancer) which are genes associated with apoptosis and cell invasion, a member of the SMAD group called “mothers against decantaplegic homolog”, and TP53 (tumor protein 53). In recent years, methylation studies of CRC have been proposed with multiple candidate genes including p16, MGMT (methylguanidine-DNA methyltransferase gene), RASSF2A (Ras association [RalGDS/AF-6] domain family member 2nd), APC, RUNX3 (Runt-related transcription factor 3 gene) and IGF2 (insulin growth factor type 2). 13 These studies have found that for most CRCs, DNA methylation analysis has found associations of methylating phenotypes with specific groups of tumors. These advances have been possible thanks to the emergence of new methodologies in the field of research. 14,15
Studies of gene methylation of promoter areas have at least four possible clinical applications: identification of tumor cells in biological samples, methylation of individual genes as prognostic markers, markers of response to chemotherapy or hormone therapy, and reactivation of genes inactivated by methylation through the use of demethylating drugs. 16
The possibility of reversing DNA methylation and reactivating genes that were affected is an attractive option as a new therapy for treatment of cancer or preneoplastic lesions, 17,18 so study of gene methylation in CRC tumors is likely to focus on identification of gene targets that could be useful for tumor analysis.
The objective of this study was to analyze the frequency of methylation of five tumor suppressor genes mentioned in other studies because they are involved in carcinogenesis in patients diagnosed with CRC and because they relate to the various clinical stages of these tumors.
Materials and method
Because previous studies have indicated the veracity of data obtained from genomic DNA extracted from peripheral blood samples this method was chosen for study of 30 patients who were undergoing surgery due to diagnoses of sporadic CRC. 19,20 Samples were taken prior to administration of patients’ first cycle of chemotherapy at the Onco-America chemotherapy center in the state of Zulia, Venezuela. Patients were at different stages of evolution, their ages varied, and both male and female patients were included. The protocol was approved by the ethics committee of the Instituto de Investigaciones Genéticas (IIG- Institute of Genetic Research) and all the patients signed a consent form prepared for this purpose. DNA was extracted using a technique developed in the Molecular Genetics Laboratory of the IIG that combines the Fenol/Sevag method and the inorganic Salting Out method. 21,22
The integrity of the DNA was confirmed through 1% agarose gel electrophoresis in TBE buffer (tris, borate and ethylenediaminetetraacetic acid) 1X at 100 V. The gel was stained with ethidium bromide. Subsequently, methylation-specific PCR (MSP) was carried out by polymerase chain reaction modified with sodium bisulfite (Promega). 19
We selected tumor suppressor genes from various metabolic pathways related to the cell cycle. They included cyclin dependent kinase inhibitor 2B (CDKN2B; p15), cyclin dependent kinase inhibitor 2A (CDKN2A; p16), an inhibitor of β-catenins, APC, the hMLH1 gene (probably the most important repairer of genomic DNA related to microsatellite instability), and the CDH1 gene (Caderina E) which encodes a transmembrane protein that participates in the complex of adhesion molecules involved in metastasis and tumor invasion. 8,9
The methyledge bisulfite conversion system kit (Promega) was used for MSP with sodium bisulfite. 19. For the study of specific methylation, the primers presented in Table 1 were used.

Subsequently, reagents contained in the kit were used to remove sulfur, and modified products were then amplified. Reactions were carried out with approximately 100 ng of modified genomic DNA, 0.2 μM of each primer, 200 μM of each of the deoxynucleotide triphosphates, 1.5 mM of magnesium chloride (MgCl) and 0.75 U of Taq polymerase (Promega) in a final volume of 25 μL. 100 ng of unmodified genomic DNA was used as a negative control. Commercial genomic DNA (Promega) methylated with SssI (Biolab) was used as the positive methylation control.
PCR products were visualized by electrophoresis of 2% agarose gel in TAE buffer (tris, acetic acid, and ethylenediaminetetraacetic acid) stained with ethidium bromide (Figures 1, 2 and 3).
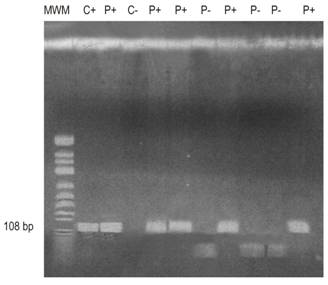
Figure 1 APC gene run shows the molecular weight marker (MWM) in column 1; the positive control (C +) in column 2; positive patients (P +) in columns lanes 3, 5, 6, 8 and 11; the negative control (C-) in column 4; and negative patients (P-) in columns 7, 9 and 10. bp: base pairs.
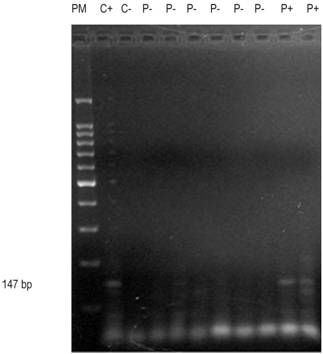
Figure 2 Agarose gel run of the P15 gene shows molecular weight marker in Column 1; control (+) in column 2; control (-) in column 3; patients (-) in columns 4-9; and patients (+) in columns 10 and 11.
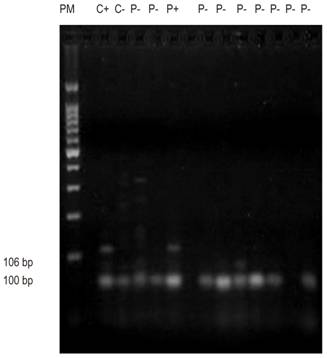
Figure 3 Agarose gel run of the E-cadherin gene shows molecular weight marker of 100 bp, positive control (C +), negative control (C-), positive patient (P +), and negative patients (P-).
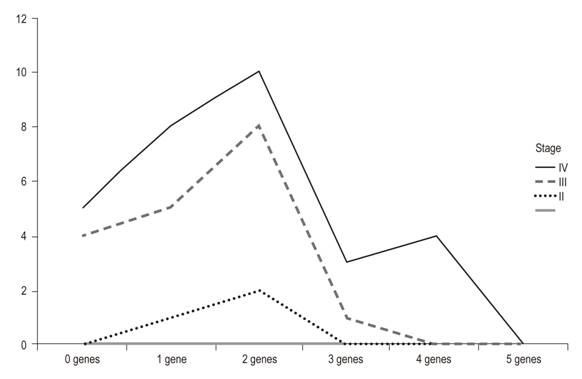
The methylation index was used as an indicator of the proportion of methylated promoter regions with respect to all of the genes studied. It was calculated by dividing the number of methylated promoter regions by the number of genes analyzed. The methylated products were grouped into tables and analyzed on the basis of percentages of methylation according to the type of gene analyzed and the stage of the tumor (Figure 4).
Results
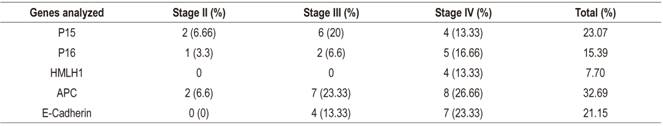
Table 2 shows clinical characteristics and methylation stages of the 30 patients studied. The average age of the patients was 60 years, and twenty out of the thirty patients were men (66.66%). The majority of the tumors (90%) were in stages III and IV of Dukes’ classification. The gene methylation statuses of the promoter regions of the genes studied (Table 3) ranged between 7.70% for hMLH1 and 32.89% for APC. In general, the methylation frequencies of the genes analyzed were similar to the averages found for the particular organ. Among the most notable differences are the high levels of methylation of the E-cadherin gene in stages III and IV. On one hand, the hMLH1 gene was observed to be 7.7% methylated in stage IV patients (Table 3), but on the other hand, a higher rate of methylation was observed in patients who had at least one methylated gene and who were in stage IV of CRC (Figure 1). Nevertheless, differences were observed in the methylation statuses of various genes in relation to the tumor stage (Table 3).
Table 2 Clinical characteristics and methylation status of genes analyzed
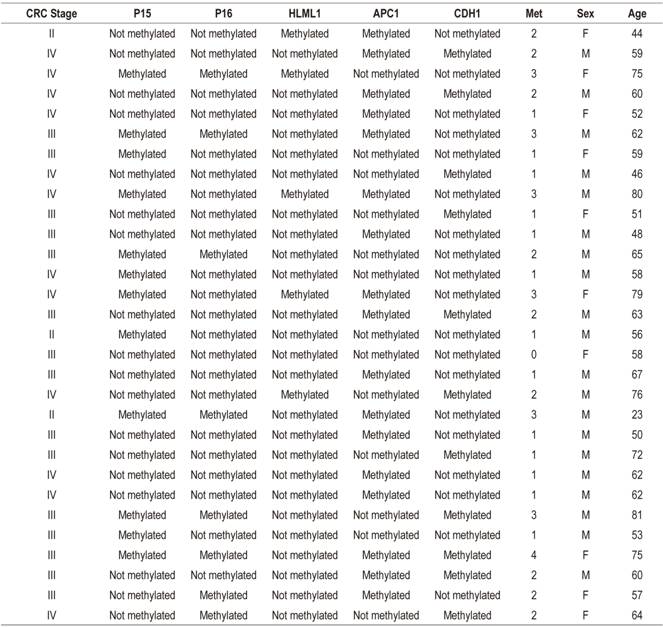
F: female; M: male; Met: number of methylated genes.
Discussion
The distribution of methylation patterns is specific to each cell type and is established during embryonic development. This alteration of DNA has been described in several diseases and in many types of cancer. In most cases, alterations may be due to hypermethylation that leads to repression of gene transcription. 9 In recent years, interest in the study of DNA methylation in CRC has been increasing. Around 50 genes related to methylation have been described in different stages of CRC making them potential biomarkers. 8,23,24
Our study analyzed the hypermethylation status of 5 glutathione S-transferase (GST) at different stages of carcinogenesis for different cancers by using MSP which established that detection of methylation can be used as a biomarker for CRC. 24
The APC gene had highest frequency of methylation with 32.89% (Figure 1) and the highest at all clinical stages (Table 3). Kudryavtserva et al. have noted that the absence of normal functioning of the APC gene results in the accumulation of β-catenins with subsequent induction of proliferation. 4 Our findings corroborate this and indicate that APC could be involved in all events in CRC from initiation and proliferation through to metastasis. APC has been proposed by other researchers as an initiator of the chromosomal instability pathway. 23,24,25
The methylation frequency of the P15 gene was second in our series, but the methylation of its promoter has been detected in very few series. Almost no methylation has been detected in this gene in series of colorectal tumors in which methylation of P15 has been analyzed. In contrast, we found a methylation rate of 23.07% (Figure 2) which is a high rate in relation to studies of the Caucasian population which observed an absence of methylation (Figure 2). However, studies of Asians have detected quite high methylation frequencies (26.1% and 68%). 26,27 In this case, the geographic origin of the patients could also explain the difference between the frequencies observed in the studies cited since our work was carried out in a mestizo population. 28
The CDH1 or E-cadherin gene’s frequency was third with 21.15% (Figure 3). This is another GST whose promoter methylation has been observed in several neoplasms including breast, lung and nasopharyngeal cancer. 15,28,29 In our CRC series the average methylation frequency was ower than that reported by Wheeler and collaborators who detected a methylation rate in CDH1 of 36%. 30 Methylation of this gene has been considered responsible for reducing the expression of E-cadherin. This gene has been involved in the appearance of metastasis which coincides with our observati0ons of stags III and IV patients. Other studies have not detected any relationship between the alteration of this gene and colorectal carcinogenesis (Xu et al., 2004; Kim et al., 2006) 31,32. Nevertheless, sample sizes could be the origin of high and discrepant observed frequencies.
GST p16, one of the most frequently altered genes in human neoplasms, was fourth in frequency in our series. 26 The methylation rate detected was 15.39%. This is low compared to findings of other studies whose rates are as high as 40% in sporadic colorectal tumors, 3,27 but it is similar to those of Van Rijnsoever et al. and Ishiguro et al. 26,33 Most studies examine patients who have undergone surgical resection of sporadic colorectal adenocarcinomas. The exceptions are the studies by Esteller and Toyota. 15,27,34 The former analyzed samples from a tumor bank whose provenances were known and which had heterogeneous characteristics. The frequency of methylation of p16 detected was 35%. In the second, methylation was analyzed along cell lines established from CRC adenocarcinomas. These were characterized by high methylation rates. The methylation frequency found in p16 was 35.39%. These studies, except for the work of Toyota and collaborators,27 analyzed the same region of the p16 promoter using MSP and used the primers described by Herman et al. 35 We also used these primers in this study. The technique used is MCA (Methylated CpG Island Amplification) which uses restriction enzymes and hybridization. The main difference between these studies and ours is the geographical origin of the patients analyzed. 28
The hMLH1 gene was fifth in frequency (13.30%, 4/30) and was only observed in stage IV of the disease. The frequencies of methylation of this gene are presented heterogeneously among publications. A study of sporadic gastric tumors in 46 patients in northern Brazil that analysed methylation of several genes found an average frequency of methylation of the hMLH1 gene of 21.74%.32,36,37 In 2013, Xia et al. pointed out that methylation of this gene is heterogeneous. They found 33% in non-selected tumors, but when they separated sporadic tumors out, they found a frequency of 16.4% which is similar to that found in our study of 2013.38 Nevertheless, they found no significant differences in relation to tumor stages. In 2013, Zhang et al. performed a metaanalysis on the prevalence of somatic mutations in the hMLh1 gene in CRC from December 1993 to September 2010.39 They concluded that this gene is mainly related to hereditary CRC and is mainly susceptible to mutations.
The use of epigenetic targets for the development of new chemotherapy treatments is emerging as a strategy with great potential since, in principle, epigenetic alterations are potentially reversible. These agents could modify patterns of DNA methylation or histone status in tumor cells through alteration of activity of enzymes responsible for the establishment and maintenance of these patterns. The drugs which have been most studied are demethylating agents which work by deactivating DNA methyltransferases (DNMT). These enzymes are responsible for transferring the methyl group to the 5-carbon of the cytosine nucleotide. Clinical studies are evaluating the use of low doses of DNMT inhibitors for various cancers including colon cancer in order to minimize toxicity. 40
Referencias
1. Agencia EFE. Cáncer colorrectal causa 49.000 muertes al año en Latinoamérica y va al alza. EFE [internet] 2018 [acceso el 20 de junio de 2018]. Disponible en: Disponible en: https://www.efe.com/efe/america/sociedad/cancer-colorrectal-causa-49-000-muertes-al-ano-en-latinoamerica-y-va-alza/20000013-3569151 . [ Links ]
2. Capote L. Resumen de las estadísticas de cáncer en el año 201. [internet] 2012 [acceso el 28 de noviembre de 2016]. Disponible en: Disponible en: http://www.oncologia.org.ve/site/userfiles/svo/Estad%C3%ADsticas%20de%20c%C3%A1ncer%20en%20el%202012.pdf . [ Links ]
3. Villalta D, Sajo A, Ovalle P. Pronósticos de la mortalidad e incidencia de cáncer en Venezuela año 2016. Caracas: Sociedad Anticancerosa de Venezuela y Centro de Estadística y Software Matemático (CESMa/USB); 2016. [ Links ]
4. Kudryavtseva AV, Lipatova AV, Zaretsky AR, Moskalev AA, Fedorova MS, Rasskazova AS, et al. Important molecular genetic markers of colorectal cancer. Oncotarget. 2016;7(33):53959-83. doi: 10.18632/oncotarget.9796. [ Links ]
5. Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers (Basel). 2013;5(2):676-713. doi: 10.3390/cancers5020676. [ Links ]
6. Bannura G, Cumsille M, Contreras J, Melo C, Barrera A, Reinero M, et al. Factores pronósticos en el cáncer colorrectal: Análisis multivariado de 224 pacientes. Rev Med Chile. 2001;129(3):237-46. doi: 10.4067/S0034-98872001000300001. [ Links ]
7. Lech G, Słotwiński R, Słodkowski M, Krasnodębski I. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J Gastroenterol. 2016;22(5):1745-55. doi: 10.3748/wjg.v22.i5.1745. [ Links ]
8. Coppedè F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Letters. 2014;342(2):238-47. doi: 10.1016/j.canlet.2011.12.030. [ Links ]
9. Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148-59. doi: 10.1056/NEJMra072067. [ Links ]
10. Doerfler W. DNA methylation and gene activity. Annu Rev Biochem. 1983;52: 93-124. doi: 10.1146/annurev.bi.52.070183.000521. [ Links ]
11. Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395-402. doi: 10.1093/hmg/9.16.2395. [ Links ]
12. Holliday R. Epigenetics: a historical overview. Epigenetics. 2006;1(2):76-80. doi: 10.4161/epi.1.2.2762. [ Links ]
13. Harrison S, Benziger H. The molecular biology of colorectal carcinoma and its implications: a review, Surgeon 2011;9(4):200-10. doi: 10.1016/j.surge.2011.01.011. [ Links ]
14. Palacio Rúa KA, Muñetón Peña CM. Bases Moleculares del cáncer colorrectal. IATREIA. 2012;25:137-48. [ Links ]
15. Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61(8):3225-9. [ Links ]
16. Roa JC. Genetic Methylation in carcinogenesis and its application in oncology. Med Clin (Barc). 2006;126(12):455-6. [ Links ]
17. Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: the time is now. Crit Rev Onco Hematol. 2008,68(11):1-11. doi: 10.1016/j.critrevonc.2008.03.001. [ Links ]
18. Szyf M. The role of DNA hypermethylation and demethylation in cancer and cancer therapy. Curr Oncol. 2008;15(2):72-5. [ Links ]
19. Mitchell S, Ross J, Drew H, Ho T, Brown G, Saunders N, et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer. 2014;14:54. doi: 10.1186/1471-2407-14-54. [ Links ]
20. Luo X, Huang R, Sun H, Liu Y, Bi H, Li J, et al. Methylation of a panel of genes in peripheral blood leukocytes is associated with colorectal cancer. Sci Rep. 2016;6:29922. doi: 10.1038/srep29922. [ Links ]
21. Miller SA, Dykes DD, Polesky HF. A simple salting-out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. doi: 10.1093/nar/16.3.1215. [ Links ]
22. Methyl edge bisulfite conversion system. Promega [internet] [acceso el 10 de abril de 2017]. Disponible en: Disponible en: http://worldwide.promega.com/~/media/Files/Resources/Protocols/Technical%20Manuals/101/MethyEdge%20Bisulfite%20Conversion%20System%20Protocol.pdf . [ Links ]
23. Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nature cell biology. 2001;3(4):433-8. doi: 10.1038/35070129. [ Links ]
24. Aoki K, Aoki M, Sugai M, Harada N, Miyoshi H, Tsukamoto T, et al. Chromosomal instability by beta-catenin/TCF transcription in APC or beta-catenin mutant cells. Oncogene. 2007;26(24):3511-20. doi: 10.1038/sj.onc.1210141. [ Links ]
25. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138(6):2059-72. doi: 10.1053/j.gastro.2009.12.065. [ Links ]
26. Ishiguro A, Takahata T, Saito M, Yoshiya G, Tamura T, Sasaki M, et al. Influence of methylated p15 INK4b and p16 INK4a genes on clinicopathological features in colorectal cancer. J Gastroenterol Hepatol. 2006;21(8):1334-9. doi: 10.1111/j.1440-1746.2006.04137.x. [ Links ]
27. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U.S.A. 1999;96(15):8681-6. doi: 10.1073/pnas.96.15.8681. [ Links ]
28. Berger M, Yang D, Sanakawa Y, Zhang W, Ning Y, Matsusaka S, et al. Impact of sex, age, and ethnicity/race on the survival of patients with rectal cancer in the United States from 1988 to 2012. Oncotarget. 2016;7(33):53668-78. doi: 10.18632/oncotarget.10696. [ Links ]
29. Mahon G, Metzger B, Dicato M. Distribution of CDH13, CDH1, MGMT and PTEN promoter methylation and KRAS, BRAF and EGFR mutation in colorectal cancer. J Clin Oncol. 2016;34(15 Suppl):e15073. doi: 10.1200/JCO.2016.34.15_suppl.e15073. [ Links ]
30. Wheeler J, Kim H, Efstathiou J, Ilyas M, Mortensen N, Bodmer W. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cáncer. Gut. 2001;48(3):367-71. [ Links ]
31. Xu X, Jian Y, Zhang H, Sun M, Jun G, Xiang D, et al. Methylation profile of the promoter CpG islands of 31 genes that may contribute to colorectal carcinogenesis. World J Gastroenterol. 2004;10(23):3441-54. doi: 10.3748/wjg.v10.i23.3441. [ Links ]
32. Kim J, Rhee Y, Bae J, Kwon HJ, Cho NY, Kim MJ, et al. Subsets of microsatellite-unstable colorectal cancers exhibit discordance between the CpG island methylator phenotype and MLH1 methylation status. Mod Pathol. 2013;26(7):1013-22. doi: 10.1038/modpathol.2012.241. [ Links ]
33. Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin. Cancer Res. 2003;9(8):2898-903. [ Links ]
34. Goto T, Mizukami H, Shirahata A, Sakata M, Ishibashi M, Kigawa M, et al. Aberrant methylation of the p16 gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2009;29(1):275-7. [ Links ]
35. Herman JC, Baylin SB. Gene Silencing in cancer in association with promoter hipermethylation. N Eng J Med, 2003;349(21):2042-54. doi: 10.1056/NEJMra023075. [ Links ]
36. Moura Lima E, Ferreira Leal M, Cardoso Smith Mde A, Rodríguez Burbano R, Pimentel de Assumpção P, Bello MJ, et al. DNA mismatch repair gene methylation in gastric cancer in individuals from northern Brazil. Biocell. 2008;32(3):237-43. [ Links ]
37. Michailidi C, Theocharis S, Tsourouflis G, Pletsa G, Kouraklis G, Patsouris E, et al. Expression and promoter methylation status of hMLH1, MGMT, APC, and CDH1 genes in patients with colon adenocarcinoma. Exp Biol Med (Maywood). 2015;240(12):1599-605. doi: 10.1177/1535370215583800. [ Links ]
38. Li X, Yao X, Wang Y, Hu F, Wang F, Jiang L, et al. MLH1 promoter methylation frequency in colorectal cancer patients and related clinicopathological and molecular features. PLoS One. 2013;8(3):e59064. doi: 10.1371/journal.pone.0059064. [ Links ]
39. Zhang R, Qin W, Xu GL, Zeng FF, Li CX. A meta-analysis of the prevalence of somatic mutations in the hMLH1 and hMSH2 genes in colorectal cancer. Colorectal Dis. 2012;14(3):e80-9. doi: 10.1111/j.1463-1318.2011.02858.x. [ Links ]
40. Lastrebner M, Flores A, Benasayag S. Tratamiento hipometilante de los síndromes mielodisplásicos. De la fisiopatogenia y la farmacología a la práctica clínica. Rev Arg Hematol. 2009;13(1):27-39. [ Links ]
Received: March 15, 2018; Accepted: June 28, 2018
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
