











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El cáncer colorrectal (CCR) consiste en el crecimiento descontrolado de las células de la mucosa de estas regiones intestinales. Es considerado uno de los mayores problemas de salud pública en muchos países desarrollados. Es la segunda causa de cáncer en los Estados Unidos, lo cual parece controversial si se considera que es curable cuando se diagnostica a tiempo. La agencia EFE en Bogotá en 2018 1 señaló que este tipo de cáncer ocasiona aproximadamente 49 000 casos al año, señalando además que las mayores tasas de Latinoamérica las presentan Uruguay (29,5 nuevos casos al año por cada 100 000 habitantes), Argentina (23,8/100 000), Cuba (19,7/100 000), Costa Rica (16,4/100 000), Brasil (15,8/100 000) y Chile (15/100 000); mientras que Guatemala (4,3/100 000) y Honduras (6,9/100 000) reportan las más bajas. En una franja intermedia se encuentran Colombia (12,9/100 000), Panamá (12,5/100 000), Paraguay (12,1/100 000), Perú (11,1/100 000), Ecuador (10,7/100 000), República Dominicana (10,2/100 000), Bolivia (9,1/100 000), Nicaragua (7,9/100 000) y México (7,8/100 000). En Venezuela, de acuerdo con las estadísticas de mortalidad del Ministerio del Poder Popular para la Salud (MPPS) en 2012, se registraron en hombres 1474 casos con 756 defunciones y para las mujeres, 1661 casos y 801 defunciones 2. En 2015 se diagnosticaron alrededor de 3500 casos en todo el país, según estadísticas de la Sociedad Venezolana de Oncología 3. El desarrollo del CCR se caracteriza por la desregulación genética y epigenética de las señales que ocurren como una cascada de transducciones a manera de multipasos cuya acumulación a largo plazo produce cambios epigenéticos que conducen a la aparición del CCR 4.
El estadio clínico-patológico es la mejor manera para predecir el curso de la enfermedad; sin embargo, este sistema ofrece solo estimaciones brutas que conducen al tratamiento innecesario de un gran número de pacientes, por una parte; y recurrencia de la enfermedad entre pacientes que solo recibieron cirugía, por otra parte 5. Junto con el hecho de que el riesgo de CCR aumenta con la edad y teniendo en cuenta que la población mundial está creciendo y envejeciendo, las próximas décadas pondrán una presión sin precedentes sobre las instituciones de atención en salud en el mundo. Por tanto, es necesario identificar biomarcadores moleculares para guiar las decisiones clínicas sobre cómo estratificar a los pacientes para regímenes de tratamiento óptimo; en particular, los pacientes con CCR en estadio II, a quienes no se les ofrece habitualmente terapia adyuvante, aunque alrededor del 20 %-30 % de ellos experimenta recaídas y muere en los 5 años posteriores a la cirugía; por su parte, los pacientes en estadio III mayores de 75 años no reciben terapia adyuvante de forma rutinaria, aunque la evidencia sugiere que existe un beneficio de tal tratamiento (6.
Los biomarcadores pronósticos que distinguen entre pacientes de alto riesgo y bajo riesgo en estas etapas están altamente justificados. La regulación epigenética de la función génica juega un papel relevante en el desarrollo y la metilación es el mecanismo que asegura la expresión específica de tipos celulares de un limitado grupo o espectro de genes 7. Diferentes enfermedades, entre ellas el cáncer, presentan patrones alterados de metilación que cambian el balance epigenético perfecto que existe en las células normales 8). La metilación del ácido desoxirribonucleico (ADN) es un proceso dinámico eficientemente regulado, en el que las secuencias no metiladas pueden ser metiladas y los grupos metilo pueden perderse, por lo que los patrones de metilación de las células somáticas son el resultado de ambas actividades: metilación y desmetilación. La reacción de metilación del ADN está catalizada por las ADN metiltransferasas e involucra la transferencia del grupo metilo de la S-adenosil-L-metionina al carbono 5 de la citosina (9. Se han identificado 3 enzimas diferentes que llevan a cabo esta reacción conocidas como las ADN metiltransferasas de mantenimiento (DNMT1) y las metilasas de novo (DNMT3A y DNMT3B). En el ser humano, las ADN metiltransferasas con actividad de novo, DNMT3A y DNMT3B, adicionan un grupo metilo a la citosina del dinucleótido CpG no metilado, creando un nuevo CpG altamente hemimetilado 10.
Este patrón aberrante de metilación está caracterizado por una hipometilación global del ADN genómico y por hipermetilación de las áreas promotoras. Tales procesos conducen a inestabilidad genómica y al silenciamiento génico, respectivamente 11,12. La represión transcripcional asociada con metilación aberrante de los islotes CpG en áreas promotoras es la consecuencia de intensos cambios de la estructura de la cromatina, producida por la interacción de metilcitosinas con diferentes complejos proteicos que reclutan enzimas que modifican histonas, como las acetilasas y metilasas de histonas 12.
Genes involucrados en diferentes vías metabólicas pueden sufrir metilación aberrante, incluidos los genes reparadores de ADN (human mut homologue 1, hMLH1), reguladores del ciclo celular como el APC (adenomatous poliposis coli), genes asociados con apoptosis e invasión celular, APC, DCC (deleted in colorectal cancer), SMAD (mothers against decantaplegic homolog) y TP53 (proteína tumoral 53), entre otros. En los últimos años se han propuesto estudios de metilación en CCR con múltiples genes candidatos, entre ellos p16, MGMT (metilguanidina-ADN metiltransferasa), RASSF2A (Ras association [RalGDS/AF-6] domain family member 2ª), APC, RUNX3 (factor de transcripción 3 relacionado con Runt) e IGF2 (factor de crecimiento insulínico tipo 2) 13. En estos trabajos se ha encontrado que en la mayoría de los CCR, los análisis de la metilación del ADN han permitido asociar fenotipos metiladores con grupos específicos de tumores. Estos avances han sido posibles gracias a la aparición de nuevas metodologías en el campo de la investigación 14,15.
El estudio de metilación génica de las áreas promotoras tiene al menos 4 posibles aplicaciones en clínica 16. Identificar células tumorales en muestras biológicas, como marcador pronóstico mediante metilación de genes individuales, marcadores de respuesta a quimioterapia u hormonoterapia, y reactivación de genes inactivados por metilación mediante el uso de fármacos demetilantes.
La posibilidad de revertir la metilación del ADN y reactivar aquellos genes afectados es una atractiva opción para su uso como un nuevo blanco terapéutico en el tratamiento del cáncer o de lesiones preneoplásicas 17,18. Es por esto que el estudio de la metilación génica en los tumores de CCR permitirá, probablemente, identificar blancos génicos con diferentes utilidades en el análisis tumoral.
El objetivo de este estudio fue analizar la frecuencia de metilación de 5 genes supresores tumorales mencionados en otras investigaciones por estar involucrados en la carcinogénesis de pacientes con diagnóstico de CCR y relacionarlos con el estadio clínico presente en ese tumor.
Material y método
Se extrajo el ADN genómico de muestras de sangre periférica, ya que estudios previos han señalado la veracidad de los datos obtenidos con este tipo de muestra 19,20 a 30 pacientes intervenidos quirúrgicamente con diagnóstico de CCR esporádico, antes de recibir su primer ciclo de quimioterapia en el centro para quimioterapias Onco-América del estado Zulia, Venezuela, en diferentes etapas de evolución, de diferentes sexos y edades. El protocolo fue aprobado por el comité de ética del Instituto de Investigaciones Genéticas (IIG) y todos los pacientes firmaron el consentimiento elaborado para tal fin. Para la extracción del ADN se utilizó la técnica combinada de Fenol/Sevag e inorgánica o Salting Out21,22 desarrollada en el Laboratorio de Genética Molecular del IIG.
La integridad del ADN se confirmó a través de una electroforesis en gel de agarosa al 1 %, en buffer TBE (tris, borato y ácido etilendiaminotetraacético) 1X a 100 V, tiñendo el gel con bromuro de etidio; posteriormente, se realizó la prueba de metilación específica mediante reacción en cadena de la polimerasa (PCR) (MSP) modificada con bisulfito de sodio (Promega) 19.
Para nuestro estudio, se seleccionaron genes supresores de tumores de diferentes vías metabólicas relacionados con el ciclo celular, entre ellos los inhibidores de cinasas dependientes de ciclina, el cyclin dependent kinase inhibitor 2B (CDKN2B; p15) y cyclin dependent kinase inhibitor 2A (CDKN2A; p16), un inhibidor de las β-cateninas, el APC, el gen hMLH1 probablemente el más importante de los genes reparadores de ADN genómico, relacionado con inestabilidad microsatelital; y el gen CDH1 (Caderina E), que codifica una proteína transmembrana que participa en el complejo de moléculas de adhesión involucrada en la metástasis e invasión tumoral 8,9.
Para la modificación específica del ADN mediante PCR (MSP) con bisulfito de sodio se utilizó el kit methyledge bisulfite conversion system (Promega) 19. Para el estudio de la metilación específica se utilizaron los iniciadores presentados en la Tabla 1.

Posteriormente, se realizó la desulfuración utilizando los reactivos contenidos en el kit y se procedió a la amplificación de los productos modificados. Las reacciones se realizaron con aproximadamente 100 ng de ADN genómico modificado, 0,2 µM de cada iniciador, 200 µM de cada uno de los desoxinucleótidos trifosfatos, 1,5 mM de cloruro de magnesio (MgCl) y 0,75 U de polimerasa Taq (Promega) en un volumen final de 25 µL. Se usó como control negativo 100 ng de ADN genómico sin modificar. Como control positivo de metilación se utilizó ADN genómico comercial (Promega) metilado con SssI (Biolab).
Los productos de PCR se visualizaron mediante electroforesis de gel de agarosa al 2 % en buffer TAE (tris, ácido acético, ácido etilendiaminotetraacético), teñidos con bromuro de etidio (Figuras 1, 2 y 3 ).
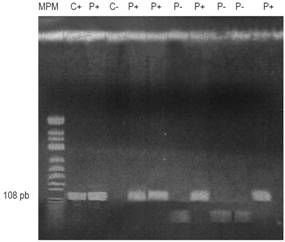
Figura 1 Corrida del gen APC muestra el carril 1: marcador de peso molecular (MPM); carril 2: control positivo (C+); carriles: 3, 5, 6, 8 y 11: pacientes positivos (P+); carril 4: control negativo (C-); carriles 7, 9 y 10: pacientes negativos (P-). pb: pares de bases.
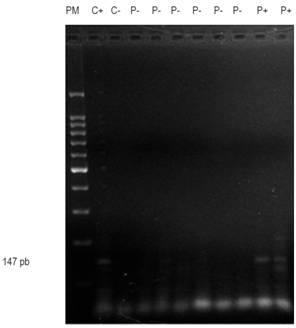
Figura 2 Gel de agarosa muestra corrida del gen P15. El carril 1 muestra marcador de peso molecular; el carril 2: control (+); carril 3: control (-); carriles 4-9: pacientes (-); carriles 10 y 11: pacientes (+).
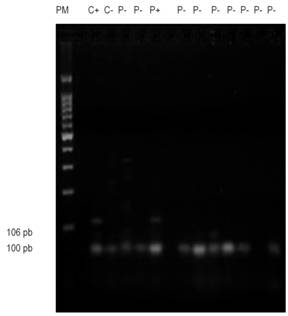
Figura 3 Gel de agarosa muestra corrida del gen E-cadherin. M: marcador de peso molecular 100 pb, C+: control positivo, C-: control negativo, P+: paciente positivo, P-: paciente negativo.
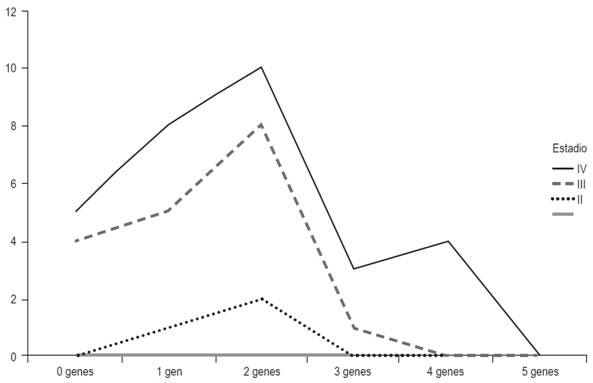
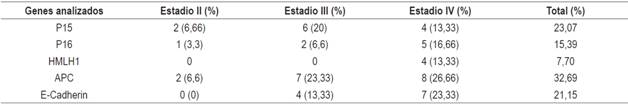
Se utilizó el índice de metilación como un indicador de la proporción de regiones promotoras metiladas respecto de la totalidad de los genes estudiados y se calculó dividiendo el número de entre el número de genes analizados, y los productos metilados se agruparon en tablas y se analizaron con base en el porcentaje de metilación según el tipo de gen analizado y el estadio del tumor (Figura 4 ).
Resultados
La Tabla 2 muestra las características clínicas y el estadio de metilación de los 30 pacientes estudiados. La edad promedio de los pacientes fue de 60 años, el sexo masculino resultó ser el más frecuentemente afectado 20 de 30 (66,66 %). La mayor parte de los tumores (90 %) se encontraba en etapas III y IV del estadio de Ducks. El estado de metilación génica de las regiones promotoras de los genes estudiados en los 30 casos de tumores CCR analizados (Tabla 3) fluctuaron entre 7,70 % para hMLH1 y 32,89 % para APC. En general, las frecuencias de metilación de los genes analizados fueron similares a los promedios encontrados para ese órgano en particular. Dentro de las diferencias más destacables se encuentra la alta metilación del gen de la E-caderina en los estadios III y IV. Por una parte, el gen hMLH1 se observó solo metilado en un 7,70 % en pacientes en estadio IV (Tabla 3). Por otra parte, se observó un mayor índice de metilación en pacientes con CCR en estadio IV en el que todos los casos presentaron al menos un gen metilado (Figura 1); sin embargo, se observaron diferencias en el estado de metilación de los diferentes genes en relación con el estadio del tumor (Tabla 3).
Tabla 2 Características clínicas y estado de metilación de los genes analizados
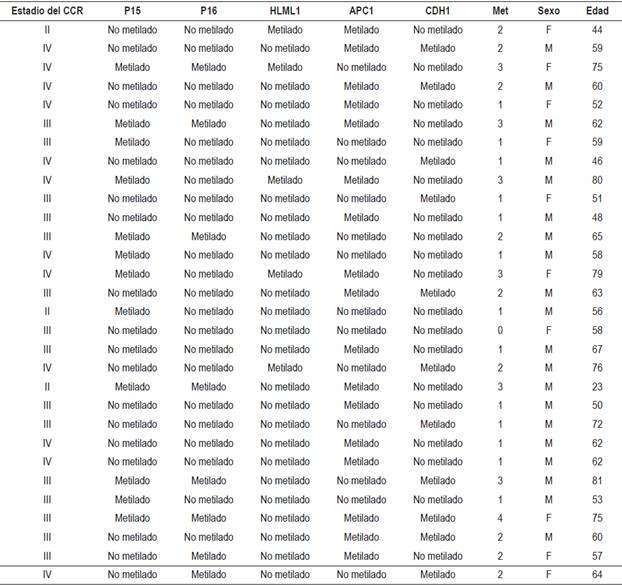
F: femenino; M: masculino; Met: número de genes metilados.
Discusión
La distribución de los patrones de metilación es específica de cada tipo celular y se establece durante el desarrollo embrionario. Esta alteración del ADN se ha descrito en varias enfermedades y en muchos tipos de cáncer. En la mayoría de estos casos la alteración puede ser por un incremento o hipermetilación que conlleva a una represión de la transcripción génica 9. En los últimos años, el interés sobre el estudio de la metilación del ADN en CCR ha ido incrementándose. Se han descrito alrededor de 50 genes relacionados con metilación en distintos estadios del CCR identificándose como potenciales biomarcadores (8,23,24.
En nuestro estudio se analizó el estado de hipermetilación de 5 glutationes S-transferasa (GST), señalados por intervenir en distintas etapas de la carcinogénesis para diferentes cánceres, mediante la utilización de PCR MSP, en cuyos reportes se establece que la detección de la metilación encontrada puede ser utilizada como biomarcador en CCR 24.
La mayor frecuencia de metilación se encontró en el gen APC con un 32,89 %, (Figura 1) y en todos los estadios clínicos presentes (Tabla 3). Kudryavtserva y colaboradores 4 señalaron que la ausencia de función normal del gen APC resulta en la acumulación de β-cateninas con posterior inducción de la proliferación; este hecho corresponde con nuestros hallazgos, en los que el APC estaría involucrado en todos los eventos tanto de inicio como de proliferación y metástasis en CCR. Esto ha sido propuesto por otros investigadores como un iniciador de la vía de inestabilidad cromosómica23,24,25.
El gen P15 fue segundo en frecuencia de metilación; la metilación de su promotor ha sido detectada en muy pocas series. Por esta razón, en algunas series de tumores colorrectales en los que se ha analizado la metilación de P15 no se ha detectado prácticamente metilación en este gen. En cuanto a nuestro trabajo, se detectó una tasa de metilación de 23,07 % (Figura 2), que es una tasa relativamente alta en relación con los trabajos concordantes con la ausencia de metilación observada en la población caucásica (Figura 2). Sin embargo, estudios en asiáticos han detectado frecuencias de metilación bastante altas (26,1 % y 68 %) 26,27. En este caso, el origen geográfico de los pacientes podría también explicar la diferencia entre las frecuencias observadas en los estudios citados, ya que nuestro trabajo se realizó en poblaciones mestizas 28.
El gen CDH1 o E-caderina fue el tercero en frecuencia con un 21,15 % (Figura 3), este es otro GST cuya metilación del promotor se ha observado en varias neoplasias, como el cáncer de mama, pulmón y nasofaríngeo15,28,29. En nuestra serie de CCR, se detectó una frecuencia media de metilación, más baja que la reportada por Wheeler y colaboradores 30 quienes detectaron una tasa de metilación en CDH1 (36 %), la metilación de este gen se ha considerado como responsable de la reducción de la expresión de la E-cadherina. Este gen ha sido involucrado con la aparición de metástasis, lo que corresponde con nuestro estudio, en el que se observó metilación en pacientes en estadios III y IV. Otros estudios no han detectado ninguna relación entre la alteración de este gen y la carcinogénesis colorrectal (Xu y colaboradores, 2004; Kim y colaboradores, 2006) 31,32. Sin embargo; el tamaño de la muestra analizada podría estar en el origen de esas altas y discrepantes frecuencias observadas.
El GST p16 fue el cuarto en frecuencia, este constituye uno de los genes más frecuentemente alterados en las neoplasias humanas 26. En nuestra serie se detectó una tasa de metilación del 15,39 %. Esta es baja en relación con los descritos en otros estudios (hasta del 40 % aproximadamente) en los tumores colorrectales esporádicos 3,27, pero similares a los de Van Rijnsoever y colaboradores 33, e Ishiguro y colaboradores 26. La mayoría de las series de tumores analizadas en estos estudios proviene de pacientes que han sido sometidos a una resección quirúrgica de adenocarcinomas colorrectales esporádicos. Las excepciones las marcan los estudios de Esteller y Toyota 15,27,34, en los que la muestra de tumores analizada en el primero proviene de un banco de tumores, con una procedencia y unas características heterogéneas, y la metilación de p16 se detectó con una frecuencia del 35 %; mientras que en el segundo, la metilación se analizó en líneas celulares, establecidas a partir de adenocarcinomas de CCR, que se caracterizan por presentar unas tasas altas de metilación, y se encontró una frecuencia de metilación en p16 del 35,39 %. La mayoría de estos trabajos analizó la misma región del promotor de p16 mediante MSP, utilizando los mismos cebadores descritos por Herman y colaboradores 35 y que se utilizaron también en el presente estudio, salvo el trabajo de Toyota y colaboradores 27, en el que la técnica utilizada es MCA (Methylated CpG Island Amplification), que consiste en la utilización de enzimas de restricción e hibridación. La principal diferencia entre estos estudios y el nuestro sería el origen geográfico de los pacientes analizados (28.
El gen hMLH1 fue el quinto en frecuencia (13,30 %; 4/30) y solo se observó en estadio IV de la enfermedad. Las frecuencias de metilación de este gen se presentan en forma heterogénea entre las diferentes publicaciones: en un estudio realizado en tumores gástricos esporádicos procedentes de 46 pacientes del norte de Brasil en el que se analizó la metilación de varios genes se detectó una frecuencia media de metilación del gen hMLH1 (21,74 %) 32,36,37. Xia y colaboradores en 2013 38 señalaron que la metilación de este gen es heterogénea, encontrando un 33 % en tumores no seleccionados, pero cuando separó los esporádicos obtuvo una frecuencia de 16,4 %, similar a la encontrada en nuestro estudio; sin embargo, no encontró diferencias significativas en relación con el estadio del tumor. Zhang y colaboradores en 2013 39 realizaron un metaanálisis sobre la prevalencia de mutaciones somáticas en el gen hMLh1 en CCR desde diciembre de 1993 hasta septiembre 2010, en el que concluyeron que este gen está principalmente relacionado con CCR hereditario y principalmente susceptible a mutaciones.
La utilización de dianas epigenéticas para el desarrollo de nuevos tratamientos de quimioterapia está emergiendo como una estrategia con gran potencial dado que, en principio, las alteraciones epigenéticas son potencialmente reversibles. Estos agentes modificarían los patrones de metilación del ADN o el estado de las histonas en las células tumorales a través de la alteración de la actividad de las enzimas responsables del establecimiento y mantenimiento de estos patrones. Los fármacos más estudiados son los demetilantes, que funcionan inactivando las metiltransferasas del ADN (DNMT). Estas enzimas son responsables de transferir el grupo metilo al carbono 5 del nucleótido citosina. Estudios clínicos están evaluando el uso de dosis bajas de inhibidores de DNMT en diferentes cánceres, incluido el de colon, para minimizar la toxicidad 40.

