










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.34 no.2 Bogotá Apr./June 2019
https://doi.org/10.22516/25007440.280
Original articles
Diagnosis and treatment of patients with hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) at a university hospital in Colombia
1Especialista en Medicina Interna, Gastroenterología y Endoscopia Digestiva, Hospital Pablo Tobón Uribe. Medellín, Colombia
2Medicina Interna y Hematología, Hospital Pablo Tobón Uribe. Medellín, Colombia
3Especialista en Medicina Interna, Universidad Pontificia Bolivariana. Medellín, Colombia
Introduction:
Hereditary hemorrhagic telangiectasia (HHT or Osler-Weber-Rendu syndrome) is a hereditary vascular disease characterized by recurrent epistaxis, gastrointestinal bleeding and chronic anemia. Many cases have arteriovenous malformations of solid organs. Diagnosis is based on clinical data, endoscopy and imaging. Early detection and treatment of complications with a multidisciplinary approach impacts the disease’s morbidity and mortality.
Objectives:
The objective of this study was to describe the demographic, clinical and outcome characteristics of patients diagnosed with HHT at a university hospital.
Results:
Records of 18 cases were obtained. The patients were from Colombia and other Caribbean countries. All diagnoses were established using the Curaçao criteria. Eleven patients 11 (61.1%) were men, and the median patient age was 56 years (IQR 52-64). The median number of hospital admissions was 6 (33.3%) (IQR 2.5-20.5), and all admissions were related to bleeding. Sixty-one percent of patients required transfusion of blood products, and the compromises of solid organs were found in the same number of patients by imaging studies.
Conclusions:
The clinical expression of THH varies, but in our study gastrointestinal manifestations were the most frequent causes of hospital admission. They frequently required transfusion of blood products and patients required multiple studies to identify the extent of the disease, and solid organ compromise. Treatment was based on endoscopic and medical management, especially administration of bevacizumab and octreotide.
Keywords: Hereditary hemorrhagic telangiectasia; hemorrhage; epistaxis; melena
Introducción:
la telangiectasia hemorrágica hereditaria (THH) es una enfermedad vascular hereditaria caracterizada por epistaxis, sangrado digestivo y anemia crónica; en muchos casos hay malformaciones arteriovenosas de órganos sólidos. El diagnóstico se realiza con base en datos clínicos, hallazgos endoscópicos e imagenológicos. La detección temprana con enfoque multidisciplinario y tratamiento de las complicaciones impacta en morbimortalidad de la enfermedad.
Objetivos:
describir las características demográficas, clínicas y desenlaces de pacientes con diagnóstico de THH en un hospital universitario.
Resultados:
se obtuvieron registros de 18 casos, 11 (61,1 %) hombres, con edad mediana de 56 años (rango intercuartílico [IQR]: 52-64). Los casos son provenientes de Colombia y algunos países caribeños. En todos los pacientes el diagnóstico se estableció mediante los criterios de Curazao. El número de ingresos hospitalarios tuvo una mediana de 6 días (IQR: 2,5-20,5). Los ingresos fueron en relación a sangrado en todos los casos, 61 % de los pacientes requirió transfusión de hemoderivados. En el 61 % de los pacientes se identificó compromiso en el órgano sólido mediante imágenes.
Conclusiones:
la THH es una enfermedad de expresión clínica variable. En nuestro estudio las manifestaciones gastrointestinales fueron las causas de ingreso más frecuentes. Se requirió con frecuencia transfusión de hemoderivados. Los pacientes requirieron múltiples estudios para identificar la extensión de la enfermedad y el compromiso de órgano sólido. El tratamiento se basó en el manejo endoscópico y médico, especialmente a base de bevacizumab y octreotida.
Palabras clave: Telangiectasia hemorrágica hereditaria; hemorragia; epistaxis; melena
Introduction
Hereditary hemorrhagic telangiectasia (HHT) or Osler-Weber-Rendu disease (OWRD) is an autosomal dominant inherited vascular disease with various clinical manifestations. Patients usually present epistaxis, gastrointestinal bleeding and iron deficiency anemia due to mucocutaneous telangiectasias. Patients with HHT are also said to be at risk of developing arteriovenous malformations which can cause serious organ damage especially in cerebral, pulmonary and hepatic circulation. 1 Initial detection of HHT is based on clinical data, and the Curacao criteria are often used (Table 1). These consist of recurrent epistaxis, telangiectasias, visceral vascular malformations and a first-degree relative with HHT (the diagnosis is established with three or more of these criteria). 2,3 Treatment of this entity consists of managing symptoms and complications. 3

The estimated prevalence of HHT is 1.5 to 2.0 people per 10,000. 4,5 Some authors think that variable penetrance (complete and incomplete) could have impact recognition and notification of the disease since not all patients present symptoms at an early age. 3,4 This entity has a higher prevalence in certain populations including the Afro-Caribbean population of Curaçao and Bonaire. 5 To date, there have been very few case series published in Latin America, but there are reports of isolated cases in which variable clinical manifestations and involvement of solid organs due to arteriovenous malformations have been described. 6,7,8
The objective of this work is to describe demographic and clinical characteristics as well as outcomes of patients diagnosed with HHT at a university hospital.
Materials and methods
This is a case series study of patients older than 18 years of age with diagnoses of HHT established in the hospital from January 2012 to July 2017. The protocol was approved by the ethics and research committee of the Hospital Pablo Tobón Uribe in Medellín, Colombia. No written authorization was required since no names, personal identity data, or photos which would allow recognition of any individuals are included in the published data. According to National Resolution 8430 of 1993, this is a minimum risk study that does not jeopardize the integrity or identity of any patients.
The review of the medical records and procedures performed on the individuals under study was carried out between January 2012 and July 2017.
Results
Records of 18 cases were obtained (Tables 2 and 3). There were 11 men (61.1%) and 9 women (29.9%) with a median age of 56 years (interquartile range [IQR]: 52-64). Six were from Antioquia (33.3%), four from Chocó (22.2%), two from Caldas (11.2%), one from Quindío (5.5%), one from Valle (5.5%), three from Bonaire (16.7%) and one from Curacao (5.5%).
Table 2 Demographic and clinical characteristics of patients with HHT.
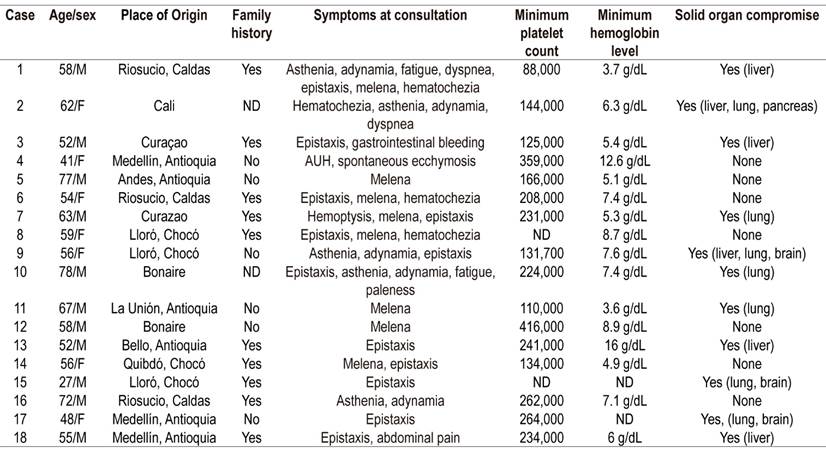
F: female, AUH: abnormal uterine hemorrhaging, M: male, ND: no data.
Table 3 Clinical data on treatment of cases with HHT
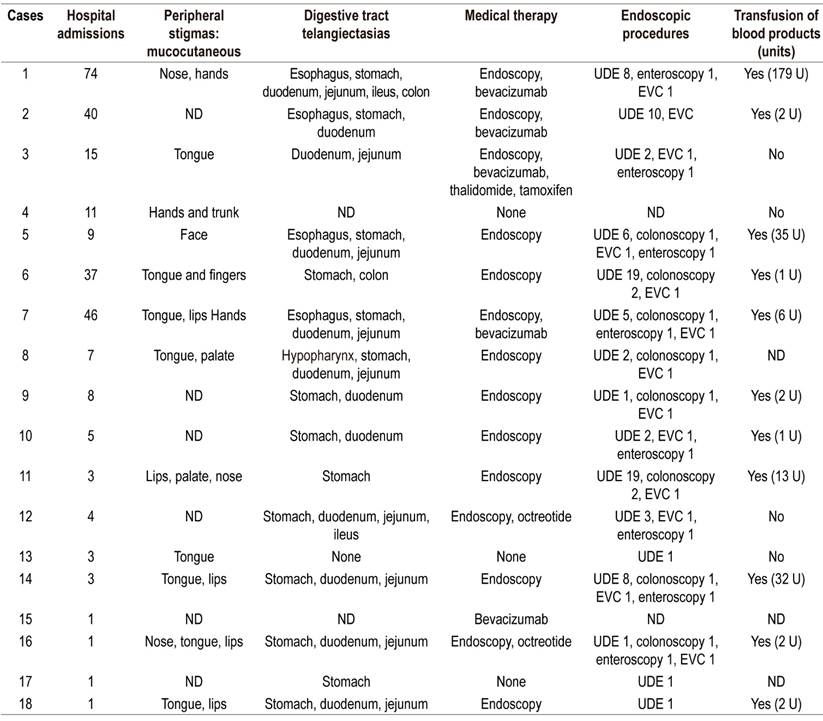
UDE: upper digestive endoscopy; U: units; EVC: endoscopic videocapsule.
First-degree family histories of HHT reported by 55.55%, six cases (33.33%) were spontaneous. There were no background data on the medical history of only two people (11.11%). The two patients from Riosucio, Caldas; one patient from Curaçao and one patient from Bonaire had first-degree family histories, and three of the four patients (75%) from Chocó had family histories of HHT.
Symptoms at admission were epistaxis (66.7%), melena (50%), hematochezia (22.22%), asthenia-adynamia (27.8%), fatigue (11.11%), dyspnea (11.11%). One patient had hemoptysis, and another patient had ecchymosis and abnormal uterine bleeding.
Endoscopy was performed and registered in the medical histories of 16 patients (88.8%), but there were no records of endoscopy in the clinical histories of the other two patients. All patients who underwent endoscopic studies had upper digestive endoscopy, endoscopic videocapsules had been used in 13/16 (81.25%), enteroscopy in 50% and colonoscopy in 50%.
Ten of the patients (55.55%) were admitted to the hospital for emergencies related to digestive bleeding manifested by melena or hematochezia. The minimum hemoglobin levels in this group of patients ranged from 3.6 to 8.9 g/dL. All endoscopic studies including video capsules were performed on these ten patients (Table 3). Seven of them (70%) required enteroscopy for argon plasma coagulation of bleeding lesions or those at risk of bleeding (Figure 1).
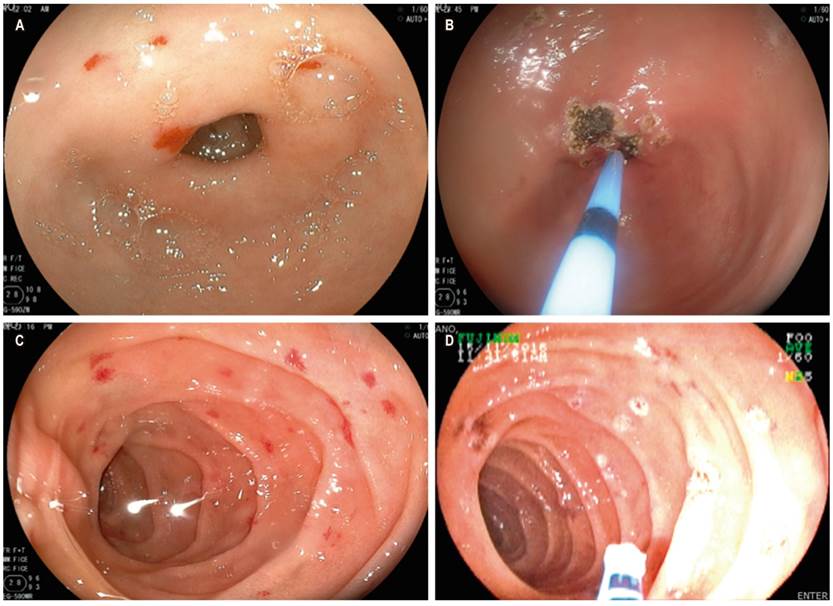
Figure 1 A. Telangiectasias in the antrum. B. Argon plasma therapy on lesions in the antrum. C. Telangiectasias in the small intestine. D. Argon plasma therapy.
Bevacizumab was administered in 27.7% of the cases, octreotide in 11.1%, thalidomide in 5.55% and tamoxifen in 5.55%.
The median hospital stay was six days (IQR: 2.5-20.5). All of these cases were patients hospitalized for digestive bleeding, and 61% of these patients required transfusion of blood products (Table 3). In 61% of these patients, solid organ involvement was identified due to arteriovenous malformations in the liver, lung, brain or pancreas. Four (22.2%) had exclusive liver compromises, three (16.7%) had exclusive lung compromises, and the others had multiorgan compromises: one patient in the liver, lung and pancreas; another patient in the liver, lung and brain; and two other patients in the lung and brain.
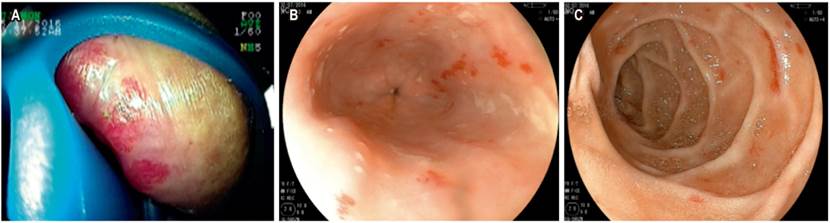
Table 2 describes the sex, age, blood test results during hospitalization (minimum platelet count and hemoglobin), and solid organ involvement due to arteriovenous malformation. Table 3 shows clinical manifestations, visible physical findings, distribution of telangiectasias in the digestive tract (Figure 2), and treatments and transfusions received.
Discussion
Most patients in our series had minor bleeding episodes at early ages that were underestimated. Several reviews describe that more than half of these patients have symptoms before 20 years of age and say that the prevalence of epistaxis may be even greater than 90% of cases.9,10 The most frequently described symptom was epistaxis. It was followed by melena, hematochezia and general symptoms of blood loss such as asthenia, adynamia, fatigue and even dyspnea. The majority had severe anemia with varied related symptoms and were in need of blood transfusions as described in a recent systematic review. 3 The number of units of red blood cells transfused had a clear relationship with the extent and severity of the disease.
In our series, no patient underwent genetic studies to determine the presence of HHT-related genes. These exams are not widely available in our environment and they are expensive. In addition, there is controversy over variable clinical expression. In a recent study, no significant differences in mortality were found in a period greater than 90 months between HHT Types 1 and 2. 11
In patients with documented telangiectasias in the digestive tract, the lesions were mostly found in proximal locations. The stomach, duodenum and jejunum were the most common sites. In most cases, EVCs were used as a non-invasive method to assess compromises in the small intestine and define the need for endoscopic therapy. These data are similar to those described in the systematic review by Jackson et al. 3 In cases where bleeding in the small intestine was evidenced, double balloon enteroscopy was used to apply argon plasma therapy, with or without systemic therapy.
Systemic therapy was used in cases with refractory bleeding. Given descriptions in the literature of increased production of vascular endothelial growth factor in patients with this entity, there could also be an imbalance between anti-angiogenic and pro-angiogenic factors. 12,13 This has allowed the use of drugs such as bevacizumab whose mechanism of action is to inhibit the growth factor of the vascular endothelium. There are multiple case reports of adults such as by Combariza et al. 14 of Hospital Pablo Tobón Uribe in Medellín. Bevacizumab has also been used in small series of cases that highlight good results in terms of effectiveness and safety.15,16 However, the costs and profile of adverse events with bevacizumab is not negligible, 17 so an appropriate patient choice must be made in order to opt for this drug. Nevertheless, we believe bevacizumab could be a promising medicine in the scenario of multiorgan involvement and refractory bleeding.
Arteriovenous malformations compromising one or more solid organs was identified in more than 60% of the cases. Most of them had pulmonary or hepatic involvement but a few also had cerebral involvement. The methods described in the literature for detecting these malformations vary. Jackson et al. found that thematic experts recommend studying pulmonary arteriovenous malformations with transthoracic contrast echocardiography and complementing it with high-resolution thoracic computed tomography if there are any abnormal findings. 3 Hepatic vascular malformations are studied in patients with confirmed HHT when they have abnormal liver function tests, cholestasis, portal hypertension or right heart failure. The study of these cases is performed by hepatic ultrasound with Doppler or three-phase helical CT.18,19,20 In our series, all cases were studied by means of magnetic resonance imaging (MRI) of the abdomen. Our hospital has good high experience with this exam, and there is less risk of nephrotoxicity than with the use iodinated contrast.
Cerebral arteriovenous alterations were identified in three patients (16.6%) which is very similar to the 10% prevalence found by Fulbright et al. with cerebral MRI. 21 This is the method most often used in asymptomatic patients with possible or confirmed HHT who are 18 years of age or older. 3
The majority of individuals with HHT who have good access to health services have normal life expectancy in relation to the general population. 3 Distribution of mortality is bimodal with peaks at 50 years and between 60 and 79 years. Acute complications related to arteriovenous malformations are the main cause of death, especially in the context of inadequate health care since these patients play a fundamental role. 3,22
We believe that more population studies are required to determine actual local prevalence: Prospective studies in which treatment alternatives aimed at reducing morbidity rates and number and durations of hospital stays are also needed as are proposals for follow-up of asymptomatic first-degree relatives.
Referencias
1. Sharathkumar AA, Shapiro A. Hereditary haemorrhagic telangiectasia. Haemophilia. 2008;14(6):1269-80. https://doi.org/10.1111/j.1365-2516.2008.01774.x [ Links ]
2. Guttmacher AE, Marchuk DA, White RI Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333(14):918-24. https://doi.org/10.1056/NEJM199510053331407 [ Links ]
3. Jackson SB, Villano NP, Benhammou JN, Lewis M, Pisegna JR, Padua D. Gastrointestinal Manifestations of Hereditary Hemorrhagic Telangiectasia (HHT): A Systematic Review of the Literature. Dig Dis Sci. 2017;62(10):2623-30. https://doi.org/10.1007/s10620-017-4719-3 [ Links ]
4. Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002;19(2):140-8. https://doi.org/10.1002/humu.10026 [ Links ]
5. Grosse SD, Boulet SL, Grant AM, Hulihan MM, Faughnan ME. The use of US health insurance data for surveillance of rare disorders: hereditary hemorrhagic telangiectasia. Genet Med. 2014;16(1):33-9. https://doi.org/10.1038/gim.2013.66 [ Links ]
6. Gómez MA, Ruiz O, Otero W. Telangiectasia hemorrágica hereditaria. Reporte de caso. Rev Col Gastrenterol 2015;30(4):469-73. https://doi.org/10.22516/25007440.11 [ Links ]
7. Sandoval DK, García E, Ramírez S, Torres KJ, Velandia MC, Villamizar JF, et al. Síndrome de Rendu Osler Weber en una adolescente en Colombia. Reporte de un caso de autopsia. Biosalud. 2018;17(1):83-9. [ Links ]
8. Giraldo A, Conde R, Varón F. Hipertensión pulmonar como manifestación de la telangiectasia hemorrágica hereditaria o síndrome de Osler-Weber-Rendú. Rev Col Neumol. 2014;26(3):139-144. https://doi.org/10.30789/rcneumologia.v26.n3.2014.39 [ Links ]
9. Alcalá-Villalón T, Castillo-González D, Agramonte-Llanes O. Enfermedad de Rendú-Osler-Weber: A propósito de 5 casos con epistaxis recurrente. Rev Cubana Hematol Inmunol Hemoter. 2012;28(3):289-98. [ Links ]
10. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48(2):73-87. https://doi.org/10.1136/jmg.2009.069013 [ Links ]
11. Kjeldsen AD, Møller TR, Brusgaard K, Vase P, Andersen PE. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J Intern Med. 2005;258(4):349-55. https://doi.org/10.1111/j.1365-2796.2005.01555.x [ Links ]
12. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997 Dec 4;390(6659):465-71. https://doi.org/10.1038/37284 [ Links ]
13. Sadick H, Riedel F, Naim R, Goessler U, Hörmann K, Hafner M, et al. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica. 2005;90(6):818-28. [ Links ]
14. Combariza JF, Olaya VP. Telangiectasia hemorrágica hereditaria. Síndrome de Osler Weber Rendú y manejo con bevacizumab. Acta Med Colomb. 2015;40:66-8. [ Links ]
15. Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;360(20):2143-4. https://doi.org/10.1056/NEJMc0901421 [ Links ]
16. Dupuis-Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307(9):948-55. https://doi.org/10.1001/jama.2012.250 [ Links ]
17. Garg N, Khunger M, Gupta A, Kumar N. Optimal management of hereditary hemorrhagic telangiectasia. J Blood Med. 2014;5:191-206. https://doi.org/10.2147/JBM.S45295 [ Links ]
18. Ravard G, Soyer P, Boudiaf M, Terem C, Abitbol M, Yeh JF, et al. Hepatic involvement in hereditary hemorrhagic telangiectasia: helical computed tomography features in 24 consecutive patients. J Comput Assist Tomogr. 2004;28(4):488-95. [ Links ]
19. Barral M, Sirol M, Placé V, Hamzi L, Borsik M, Gayat E, et al. Hepatic and pancreatic involvement in hereditary hemorrhagic telangiectasia: quantitative and qualitative evaluation with 64-section CT in asymptomatic adult patients. Eur Radiol. 2012;22(1):161-70. https://doi.org/10.1007/s00330-011-2243-y [ Links ]
20. Garcia-Tsao G, Korzenik JR, Young L, Henderson KJ, Jain D, Byrd B, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000;343(13):931-6./10.1056/NEJM200009283431305. [ Links ]
21. Fulbright RK, Chaloupka JC, Putman CM, Sze GK, Merriam MM, Lee GK, et al. MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. AJNR Am J Neuroradiol. 1998;19(3):477-84. [ Links ]
22. Sabbà C, Pasculli G, Suppressa P, D’Ovidio F, Lenato GM, Resta F, et al. Life expectancy in patients with hereditary haemorrhagic telangiectasia. QJM. 2006;99(5):327-34. https://doi.org/10.1093/qjmed/hcl037 [ Links ]
Received: August 02, 2018; Accepted: October 03, 2018
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
