











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroducción
La telangiectasia hemorrágica hereditaria (THH) o síndrome de Rendu-Osler-Weber (SOWR) es una enfermedad vascular hereditaria autosómica dominante con diferentes manifestaciones clínicas. Los pacientes suelen presentar epistaxis, hemorragia gastrointestinal y anemia por deficiencia de hierro debido a telangiectasias mucocutáneas. Se ha descrito que los pacientes con THH también corren el riesgo de desarrollar malformaciones arteriovenosas especialmente en la circulación cerebral, pulmonar y hepática, que pueden causar un grave daño orgánico 1. La detección inicial del SOWR se basa en datos clínicos y a menudo se utilizan los criterios de Curazao (Tabla 1), que consisten en epistaxis recurrente, telangiectasias, malformaciones vasculares viscerales y un familiar de primer grado con SOWR (el diagnóstico establecido se realiza con 3 o más de estos criterios) 2,3. El tratamiento de esta entidad consiste en el manejo de los síntomas y de las complicaciones 3.

La prevalencia estimada de la THH es 1,5-2 personas por 10 000 4,5. Algunos autores describen que la penetrancia variable (completa e incompleta) podría impactar en el reconocimiento de la enfermedad y en la notificación de la misma, ya que no todos los pacientes presentan síntomas en edades tempranas 3,4. Esta entidad tiene una mayor prevalencia en ciertas poblaciones, como en afrocaribeños de Curazao y Bonaire 5. Hasta el momento hay escasas series de casos publicadas en América Latina, pero sí hay reportes de casos aislados en los que se describen manifestaciones clínicas variables y también el compromiso en órganos sólidos por malformaciones arteriovenosas 6,7,8.
El objetivo de este trabajo es describir las características demográficas y clínicas y los desenlaces de pacientes con diagnóstico de THH en un hospital universitario.
Materiales y métodos
Es un estudio tipo serie de casos. Se realizó en pacientes mayores de 18 años, con diagnóstico establecido de THH o SOWR en el hospital, en el período de enero de 2012 a julio de 2017. El protocolo fue aprobado por el comité de ética e investigaciones del Hospital Pablo Tobón Uribe, Medellín, Colombia. No se requirió autorización escrita, ya que en los datos publicados no se revelaron nombres, datos personales de identidad, ni fotos que permitan reconocer al individuo. De acuerdo con la Resolución Nacional 8430 de 1993, el estudio se clasifica como de riesgo mínimo, es decir, que no pone en riesgo la integridad ni la identidad del paciente.
Se realizó la revisión de las historias clínicas y de los procedimientos realizados en los individuos a estudio en el período comprendido entre enero de 2012 y julio de 2017.
Resultados
Se obtuvieron registros de 18 casos (Tablas 2 y 3): 11 hombres (61,1 %) y 9 mujeres (29,9 %), con mediana de edad de 56 años (rango intercuartílico [IQR]: 52-64); 6 provenientes de Antioquia (33,3 %), 4 de Chocó (22,2 %), 2 de Caldas (11,2 %), 1 de Quindío (5,5 %), 1 de Valle (5,5 %), 3 de Bonaire (16,7 %) y 1 de Curazao (5,5 %).
Tabla 2 Características demográficas y clínicas de pacientes con THH
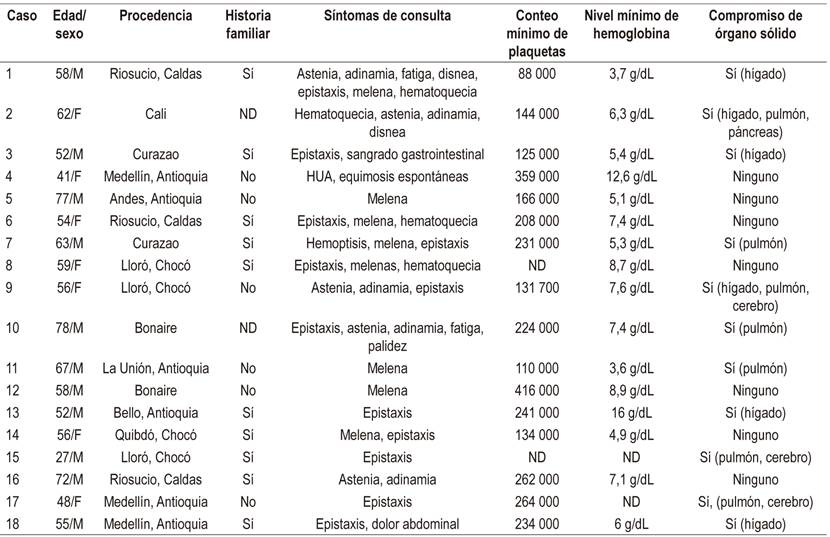
F: femenino; HUA: hemorragia uterina anormal; M: masculino; ND: no hay datos.
Tabla 3 Datos clínicos, tratamientos recibidos de los casos con THH
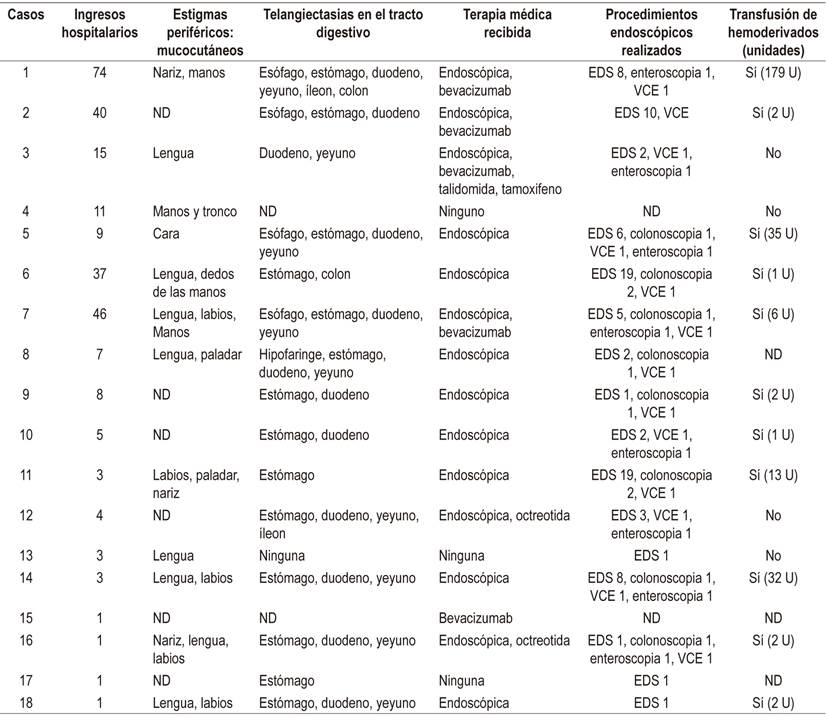
EDS: endoscopia digestiva superior; U: unidades; VCE: videocápsula endoscópica.
El 55,55 % refirió historia familiar de primer grado de THH, 6 casos (33,33 %) fueron espontáneos y en solo 2 personas (11,11 %) no había datos referentes en los antecedentes en la historia clínica. Los 2 pacientes de Riosucio, Caldas, 1 de Curazao y 1 de Bonaire tenían el antecedente familiar de primer grado; así mismo, 3/4 (75 %) de los provenientes del Chocó hicieron referencia de su historia familiar de THH.
Los síntomas referidos al momento del ingreso fueron: epistaxis en el 66,7 %, melenas en el 50 %, hematoquecia en el 22,22 %, astenia-adinamia en el 27,8 %, fatiga en el 11,11 %, disnea en el 11,11 % y otros síntomas como hemoptisis fue referido por 1 paciente; equimosis y hemorragia uterina anormal (HUA) por otro paciente.
Se realizaron estudios endoscópicos en 88,8 % de los pacientes, en el resto no se registraron estudios en la historia clínica. A todos los pacientes con estudios endoscópicos se les registró endoscopia digestiva superior, videocápsula endoscópica (VCE) en 13/16 (81,25 %), enteroscopia en el 50 % y colonoscopia también en el 50 %.
El 55,55 % de los pacientes ingresó al hospital por urgencias en relación con el sangrado digestivo manifestado por melena o hematoquecia, la hemoglobina mínima de ingreso en este grupo de pacientes estuvo en rangos de 3,6-8,9 g/dL. De estos, se les realizaron estudios endoscópicos a todos. Al 100 % de estos casos se les realizó VCE (Tabla 3), de los cuales el 70 % requirió enteroscopia con fines terapéuticos (aplicación de argón plasma sobre lesiones con potencial riesgo de sangrado o sangrantes) (Figura 1).
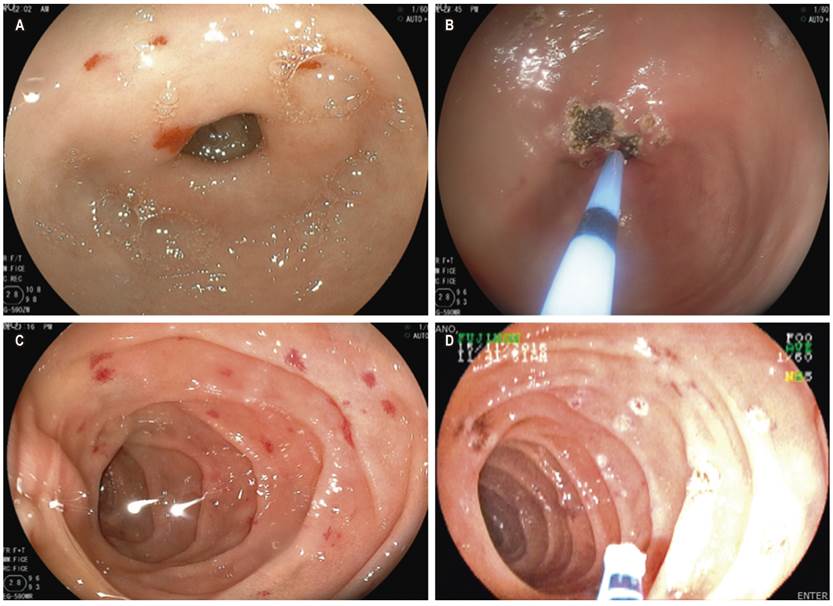
Figura 1 A. Telangiectasias en el antro. B. Terapia con argón plasma sobre las lesiones en el antro. C. Telangiectasias en el intestino delgado. D. Terapia con argón plasma.
En cuanto a la terapia médica prescrita, el bevacizumab se administró en el 27,7 % de los casos, octreotida en el 11,1 %, talidomida en el 5,55 % y tamoxifeno en el 5,55 %.
El número de ingresos hospitalarios tuvo una mediana de 6 días (IQR: 2,5-20,5), todos los casos estuvieron relacionados con sangrado digestivo; el 61 % de los pacientes requirió transfusión de hemoderivados (Tabla 3). En el 61 % de los pacientes se identificó compromiso en órgano sólido por malformaciones arteriovenosas (hígado, pulmón, cerebro o páncreas): la afectación exclusiva del hígado se presentó en 4 de ellos (22,2 %); el compromiso exclusivo del pulmón en 3 individuos (16,7 %); en el resto de pacientes el compromiso fue multiorgánico: 1 paciente en el hígado, pulmón y páncreas; en otro paciente, hígado, pulmón y cerebro; y en otros 2 pacientes, pulmón y cerebro.
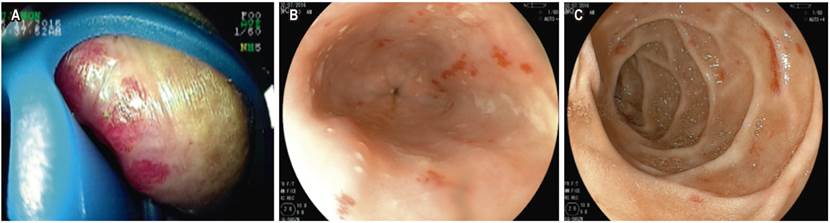
En la Tabla 2 se describe el sexo, edad, exámenes hematológicos tomados durante la hospitalización (conteo mínimo de plaquetas y de la hemoglobina) y el compromiso de órgano sólido por malformación arteriovenosa. En la Tabla 3 se anotan las manifestaciones clínicas y los hallazgos físicos visibles, además de la distribución de las telangiectasias en el tracto digestivo (Figura 2); también se describen las terapias recibidas y las transfusiones.
Discusión
En nuestra serie, la mayoría de los pacientes tuvo episodios menores de sangrado que fueron subestimados en edades tempranas. En varias revisiones se describe que más de la mitad de los pacientes presenta síntomas antes de los 20 años de edad y la prevalencia de la epistaxis puede ser incluso mayor del 90 % de los casos 9,10. Los síntomas descritos con más frecuencia fueron epistaxis, seguidos por melena, hematoquecia y síntomas generales de pérdidas hemáticas como astenia, adinamia, fatiga e incluso disnea. La mayoría tenía anemia grave, con síntomas variados en relación con esta y con necesidad de transfusión de hemoderivados, como se describió en una revisión sistemática reciente 3. El número de unidades de glóbulos rojos transfundidas tuvo una clara relación con la extensión y gravedad de la enfermedad.
En nuestra serie, a ningún paciente se le realizaron estudios genéticos para determinar la presencia de genes relacionados con la THH. Estos exámenes no están ampliamente disponibles en nuestro medio y su realización es costosa; además, existe controversia sobre la expresión clínica variable. En un estudio reciente no se encontraron diferencias significativas en la mortalidad en un período mayor de 90 meses entre THH tipos 1 y 2 11.
En los pacientes a quienes se les documentó telangiectasias en el tracto digestivo, las lesiones se encontraron mayormente en una localización proximal. El estómago, duodeno y yeyuno fueron los sitios más comunes. En la mayoría de casos se utilizó la VCE como método no invasivo para evaluar el compromiso en el intestino delgado y definir la necesidad de terapia endoscópica. Estos datos son similares a lo descrito en la revisión sistemática de Jackson y colaboradores 3. En los casos en los que se evidenció sangrado en el intestino delgado, se utilizó enteroscopia de doble balón con fines terapéuticos con aplicación de terapia argón plasma, con o sin terapia sistémica.
La terapia sistémica se utilizó en casos con sangrado refractario. Teniendo en cuenta que se ha descrito que los pacientes con esta entidad tienen aumento en la producción del factor de crecimiento del endotelio vascular, también podría haber un desbalance entre factores antiangiogénicos y proangiogénicos 12,13. Esto ha permitido utilizar medicamentos cuyo mecanismo de acción sea inhibir el factor de crecimiento del endotelio vascular como el bevacizumab. Hay múltiples reportes de casos en adultos como el descrito por Combariza y colaboradores 14 del Hospital Pablo Tobón Uribe en Medellín. También se ha usado en pequeñas series de casos en las que se resaltan buenos resultados en cuanto a la efectividad y seguridad 15,16. Los costos y el perfil de eventos adversos del medicamento no son despreciables 17, por lo que se debe hacer una adecuada elección del paciente para definir dicha terapia. No obstante, creemos que podría ser un medicamento prometedor en el escenario de compromiso multiorgánico y sangrado refractario.
El compromiso por malformaciones arteriovenosas en órgano sólido se identificó en más del 60 % de los casos, estos en su mayoría tenían compromiso pulmonar o hepático, seguidos de afectación cerebral. En este aspecto, lo descrito en la literatura es variable en cuanto a métodos utilizados para detectar dichas malformaciones. Jackson y colaboradores encontraron que los expertos temáticos recomiendan estudiar las malformaciones arteriovenosas pulmonares con ecocardiograma transtorácico contrastado y, en caso de que se encuentren hallazgos anormales, complementan con tomografía axial computarizada (TAC) torácica de alta resolución 3. Las malformaciones vasculares hepáticas se estudian en pacientes con THH confirmado cuando tengan alteración en las pruebas de función hepática, colestasis, hipertensión portal o insuficiencia cardíaca derecha. El estudio de estos casos se realiza mediante ecografía hepática con Doppler o TAC helicoidal trifásico 18,19,20. En nuestra serie, a todos los casos se les estudió mediante resonancia magnética nuclear (RMN) contrastada de abdomen; este es un examen disponible en nuestro hospital, con alta experiencia y tiene menos riesgo de nefrotoxicidad en comparación con el contraste yodado, por lo que se considera de elección en este escenario.
Se identificaron alteraciones arteriovenosas cerebrales en el 16,6 %, dato muy parecido a lo descrito por Fulbright y colaboradores, quienes mencionaron una prevalencia del 10 % con resonancia cerebral 21. La resonancia cerebral es el método más utilizado después de los 18 años en pacientes asintomáticos con THH posible o confirmada 3.
En cuanto al pronóstico, la mayoría de individuos con THH que tienen un acceso bueno a los servicios de salud tiene una expectativa de vida normal en relación con la población general 3. Hay una distribución bimodal de la mortalidad, con picos a los 50 años y entre los 60 y 79 años. Las complicaciones agudas en relación con las malformaciones arteriovenosas son la causa principal de la muerte, en especial en el contexto de un inadecuado cuidado de la salud, ya que juega un papel fundamental el seguimiento de estos pacientes 3,22.
Consideramos que se requieren más estudios poblacionales para determinar la prevalencia local real, además de estudios prospectivos en los cuales se planteen alternativas de tratamiento que impacten en menos tasas de morbilidad y menos hospitalizaciones; y también propuestas para el seguimiento de familiares de primer grado asintomáticos.

