










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista colombiana de Gastroenterología
versión impresa ISSN 0120-9957
Rev Col Gastroenterol vol.35 no.1 Bogotá ene./mar. 2020
https://doi.org/10.22516/25007440.375
Review articles
Diagnostic and therapeutic approach for cholestasis in the adult
1Médico Internista, Universidad Javeriana. Gastroenterólogo, Universidad Nacional de Colombia. Especialista en servicio de endoscopia y trasplante hepático, Fundación Santa Fe de Bogotá. Docente de la Facultad de Medicina, Universidad de los Andes. Bogotá, Colombia
2Médica internista. Hepatóloga, Fundación Santa Fe de Bogotá, Bogotá, Colombia
3Médico internista. Gastroenterólogo, Universidad Nacional de Colombia; Centro de Enfermedades Digestivas, GutMédica. Bogotá, Colombia
Cholestasis is one of the most frequent reasons for hepatology consultation. It is generated by altered synthesis, secretion or flow of bile through the biliary tract and is defined by elevated levels of enzymes such as alkaline phosphatase and gamma glutamyl transferase. In late stages, hyperbilirubinemia and clinical manifestations such as pruritus and jaundice develop. The diagnostic approach involves establishment of the reasons for elevated enzyme levels and determination of whether it is intrahepatic or extrahepatic. If it is intrahepatic, the source must be determined (hepatocytes, small bile ducts, or large caliber bile ducts). Treatment depends on the etiology, so accurate diagnosis is important. This review presents the pathophysiology and a diagnostic and therapeutic approach.
Keywords: Cholestasis; intrahepatic; extrahepatic
La colestasis es uno de los motivos de consulta más frecuentes en hepatología. Se genera por una alteración en la síntesis, la secreción o el flujo de la bilis, a través del tracto biliar. Esta se define por una elevación de enzimas como la fosfatasa alcalina (Alkaline Phosphatase, ALP) y la gamma-glutamil transferasa, y en estadios tardíos con la hiperbilirrubinemia, al igual que con otras manifestaciones clínicas, tales como el prurito y la ictericia.
El enfoque diagnóstico implica establecer el origen de dicha elevación, determinando si es intrahepática o extrahepática. Si es intrahepática, se debe esclarecer si proviene de los hepatocitos o de la vía biliar de pequeño y de gran calibre. El tratamiento dependerá de la etiología, por lo cual es importante un diagnóstico preciso. En esta revisión se presenta la fisiopatología y un enfoque diagnóstico y terapéutico.
Palabras clave: Colestasis; intrahepática; extrahepática
Introduction
The term cholestasis comes from the Greek words chole, which means bile, and stasis, which translates to still. Cholestasis is defined as the syndrome generated by alteration of synthesis, secretion and/or flow of bile through the biliary tract. It manifests initially as higher levels of serum alkaline phosphatase (ALP) levels and gamma-glutamyl transferase as well as fatigue and generalized pruritus without skin lesions. 1 Although jaundice is an important sign of cholestasis, it may be absent, particularly in adults with chronic asymptomatic cholestatic diseases. 2
Cholestasis has been classified according to cases involved in the excretion of bile related to intrahepatic etiology such as compromised hepatocellular cytoplasm and/or compromised medium-sized bile ducts (up to 400 µm in diameter) and the extrahepatic causes in large bile ducts are compromised. Causes of obstruction of the bile duct can be stones, pancreatic or biliary tumors, and hilar metastases. 3
Pathophysiology
To understand the origin of cholestasis, one must start from the composition of the liver lobule. This unit arises from the smallest functional portion of the liver in which hepatocytes are arranged in plates along the blood flow from the portal vein to the central vein. Within these plates, the hepatocytes form tubular lumens called canaliculi in which initial bile formation occurs.
These hepatocytes contain two intake and export systems located in the basolateral (sinusoidal) and canalicular (apical) portions of the membrane of hepatocytes and cholangiocytes. A process of osmosis generates a flow of water that secretes the bile from the smallest to the largest bile ducts (Figure 1). 4-6
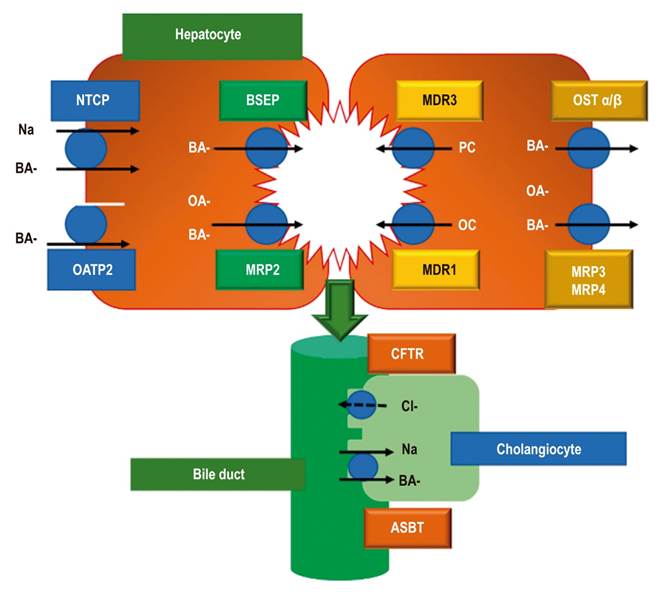
Figure 1 Bile acid transport systems. NTCP: Na-Taurocholate Cotransporting Polypeptide; OATP2: Organic Anion Transporting Polypeptide 2; BSEP: Bile Salt Export Pump; MRP2: multidrug resistance-associated protein 2; MDR3: Multidrug Resistance protein 3; MDR1: Multidrug Resistance protein 1; Organic Solute Transporter alpha and beta; MRP3: multi-drug resistance protein type 3; MRP4: multidrug resistance resistance-associated protein 4; CFTR: Cystic Fibrosis Transmembrane conductance Regulator; ASBT: Apical Sodium - Bile acid Transporter; BA: bile acid; OA: organic anion; PC: Phosphatidylcholine. Taken from reference 5.
Captured bile acids enter via Na-Taurocholate Cotransporting Polypeptide (NTCP) receptors which allow bile salts to concentrate in hepatocytes. 6-8 Bile acids also enter through Organic Anion Transporting Polypeptides (OATP2/OATP1B1) located in the basolateral membranes of hepatocytes. 5
Bile acids are excreted into the bile by the canalicular Bile Salt Export Pump (BSEP) and by the conjugated canalicular exporter protein MRP2 (Multidrug Resistance-associated Protein 2).
The MRP2 transporter (ABCC2) secretes organic anions like conjugated bilirubin, glutathione and even antibiotics like ceftriaxone. 6 Other transporters located in the canalicular membrane are MDR1 (Multidrug Resistance 1); glycoprotein P (ABCB1) which pumps organic components and cationic drugs out of the cell; and MDR3 (Multidrug Resistance 3) which excretes phospholipids such as phosphatidylcholine which, in combination with cholesterol and bile acids, forms micelles (Figure 1). 4-6
Primary bile salts, cholate and chenodeoxycholate, are formed by enzymatic modification of cholesterol which confers hydrophilic characteristics leading to the formation of micelles whose primary function is to take lipids from the intestine. 8,9 Nevertheless, they are potentially dangerous for the integrity of the cell membrane under conditions of cholestasis. 6
Meanwhile, cholangiocytes forming the bile duct provide another series of receptors that facilitate reabsorption of bile acids such as apical sodium-dependent bile acid transporter (ASBT) and CFTR (Cystic Fibrosis Transmembrane conductance Regulator) which is the transmembrane regulator of cystic fibrosis. 5
Once the bile salts have been secreted into the intestinal lumen, they are recaptured by an ASBT in the enterohepatic circulation in the ileum. 9 When they reach a hepatocyte, they are again taken up by the NTCP receptor which allows the bile salts to concentrate within the hepatocyte. 5,6 Other receptors called MRP3 (Multidrug Resistance-associated Protein 3) and MRP4 (Multidrug Resistance-associated Protein 4) and OST (Organic Solute Transporter) alpha and beta are located in the basolateral membrane of the hepatocyte. They are an alternative excretion route for bile acids and other organic anions which lead into the systemic circulation. 5
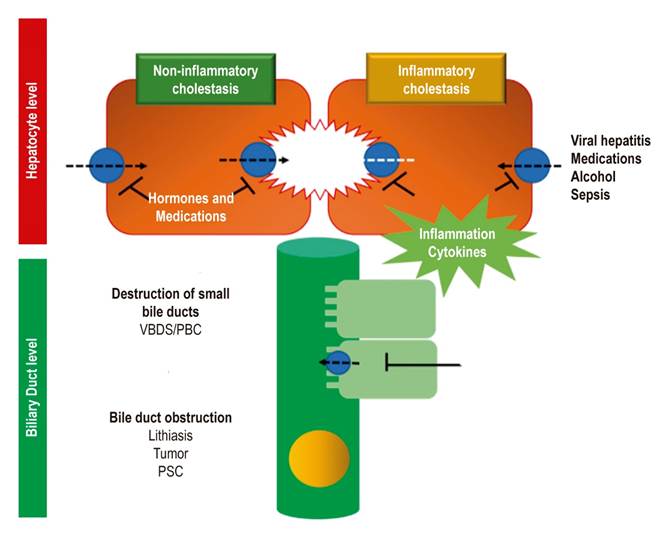
Cholestasis results from a functional defect in the formation of bile in a hepatocyte or from an alteration in its secretion and flow through the bile ducts (Figure 2). 4,5
Clinical manifestations
Cholestasis’ spectrum of symptoms is generated by accumulation of substances in the liver and blood which are generally excreted in the bile. Patients with this syndrome may be asymptomatic and sometimes report itching and jaundice. However, jaundice may be absent in adults with chronic cholestatic liver disease. 2
Steatorrhea is an important finding. It is defined as the loss of more than 10 g of fat in feces per day following consumption of 70 g/d. 10 Steatorrhea is secondary to inadequate concentration of postprandial bile in the small intestine. This causes malabsorption of fat and fat-soluble vitamins that usually help in the absorption of these elements. 3,9 Steatorrhea can be accompanied by weight loss and acropachy (also called Hippocratic fingers). 2
Fat-soluble vitamin deficiency has a wide range of neurological symptoms. They include night blindness secondary to vitamin A deficiency; hyporeflexia and/or ataxia secondary to myelopathy (vitamin E deficiency); coagulopathy secondary to vitamin K deficiency, and disorders of the musculoskeletal system such as osteomalacia, osteoporosis, and fractures secondary to vitamin D and calcium deficiency. 3,9,10 Some studies have suggested that persistent hyperbilirubinemia of more than 2-3 mg/dL is associated with fat-soluble vitamin deficiency. 11-13
In addition, an association of osteoporosis and fat-soluble vitamin deficiency with prolongation of prothrombin time has been documented. Factors 2, 7, 9 and 10 are dependent on vitamin K which cannot be absorbed during episodes of cholestasis. 14
Similarly, progression of diseases such as primary biliary cholangitis that are associated with cholestasis can continue until portal hypertension manifests as a result of ascites, encephalopathy, and upper gastrointestinal bleeding. 10 Physical examination may find xanthomas (cholesterol deposits in the tendons or bony prominences in the elbows and knees) or xanthelasmas (lipid deposits in the periorbital folds). 2,11
Paraclinical tests
The most sensitive test for identifying cholestasis is measurement of serum bile acids, but it is frequently not available. When it is not, tests for alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT) levels, which are biochemical markers of chronic cholestasis, should be used. 2
ALP levels may rise due to physiological causes. Elevation occurs in the first 3 months of life and during puberty. ALP levels gradually increase during pregnancy and between the ages of 40 and 65, especially in women. 2,10 ALP levels in male adolescents can reach 2 to 5 times the normal value for adults. This correlates with bone growth. 15
African-Americans have between 10% and 15% higher serum ALP levels than the overall population. Smokers’ ALP levels can be 10% higher than those of non-smokers. 10 Similarly, people whose blood types are O and B may also have higher than average ALP levels after a high-fat meal due to the intestinal influence of the enzyme. 2,10,11
The pathological causes that cause ALP levels to increase range from bone diseases such as fractures, Paget’s disease, and osteomalacia to vitamin D deficiency, heart failure, kidney failure, hyperthyroidism, hyperparathyroidism, hematological malignancies such as leukemia and lymphoma, and kidney cancer. 10
This phenomenon arises as a result of the presence of specific ALP isoforms in tissue as well as their location on the cell surface. ALP isoforms include the Regan isoenzyme (also called placental-like alkaline phosphatase (PLALP)), Intestinal Alkaline Phosphatase (IALP), Liver/Bone/Kidney, ALP, and Germ Cell Alkaline Phosphatase (GCALP - also known as the Nagao isoenzyme). ALP location on the cell surface hydrolyzes monophosphate esters at high pH and releases inorganic phosphate. 16-18
Whether elevated ALP levels have an intrahepatic or extrahepatic source (mainly in the bones, intestine, leukocytes and placenta) must be determined. This is done by measuring gamma-glutamyl transferase levels of ALP isoenzymes levels. 2,11 Bone activity accounts for around half of ALP in adults, making it the most important and useful isoenzyme for this study. 18
Nevertheless, gamma-glutamyl transferase’s specificity for cholestatic diseases is low since its levels are high in up to 50% of alcoholic patients without evidence of liver disease. 19 High GGT levels also occur in etiologies such as pancreatic diseases, myocardial infarcts, kidney failure, and emphysema, 20,21 and they occur in patients taking medications such as phenytoin and barbiturates.
Consequently, GGT tests should not be used for screening for underlying liver disease in the absence of abnormalities in other liver tests. 15 Conjugated serum bilirubin levels may or may not be high during cholestasis. 2,22,23
Etiology
Genetic alterations that appear in childhood, autoimmune disorders, systemic disorders, and disorders secondary to drugs area among the many causes of cholestasis. However, one way to categorize the etiology of cholestasis is to divide it into intrahepatic and extrahepatic causes. 3
Intrahepatic causes are generally caused by inflammatory and destructive conditions often referred to as vanishing bile duct syndrome (VBDS). 3,5 Primary biliary cholangitis (PBC), formerly known as primary biliary cirrhosis, is one of the most frequent autoimmune causes of cholestasis. It generates lymphocytic granulomatous cholangitis that involves small bile ducts. 3,24,25
In general, PBC occurs in women (ratio 9:1) between the ages of 40 and 60. It is accompanied by positive anti-mitochondrial antibodies (AMA) (titers> 1:40) directed against the E2 subunit of pyruvate dehydrogenase. 22 Its diagnosis is made based on the presence of at least two of three criteria: cholestasis due to high ALP levels, positive AMAs, and histopathological evidence of nonsuppurative cholangitis with destruction of medium and small caliber interlobular bile ducts. 26
In contrast, primary sclerosing cholangitis (PSC) predominates in men by a ratio of 2:1 compared to women. Its average age of onset is approximately 40 years. PSC affects the intrahepatic and extrahepatic bile ducts where it generates strictures and subsequent fibrosis and cirrhosis. 27,28 Diagnosis of PSC involves a combination of imaging of localized strictures throughout the entire biliary tree which are identifiable by endoscopic retrograde cholangiopancreatography or magnetic resonance cholangiopancreatography (MRCP). Changes in the bile duct can be seen from a liver biopsy which shows concentric periductal fibrosis (onion skin fibrosis). 27
Similarly, anti-smooth muscle antibodies (ASMA), antinuclear antibodies (ANA) and antineutrophil cytoplasmic antibodies (ANCA) can be identified in up to 50% of patients although they are not specific to PSC. 28
Other diseases with autoimmune substrates which can cause cholestasis and share the same pathophysiology of periductal fibrosis and progressive loss of bile ducts are G4 immunoglobulin cholangiopathy; chronic ductopenic rejection in liver transplantation and graft-versus-host disease. The latter appears in the first 100 days following allogeneic hematopoietic cell transplantation.
Even lymphoma, either Hodgkin’s or non-Hodgkin’s, can manifest in up to 10% of patients with jaundice and VBDS and in 40% of those with elevated ALP levels. It is accompanied by classic B symptoms such as fever and weight loss and presents a spectrum that ranges from a solitary lesion to diffuse infiltrative involvement. It responds to both chemotherapy and radiation therapy. 3,29
Drug induced liver injury (DILI)) leads to fulminant liver failure in 13% of these patients in the United States. DILI manifests with a hepatocellular pattern and high levels of transaminases, or a cholestatic pattern with ALP levels more than two times the upper limit of normal. 30
Changes generated by medications can range from slight inflammation of the parenchyma and mild ductopenia to progressive inflammation, fibrosis and the loss of bile ducts 31. Among the key drugs that triggers these symptoms are antibiotics such as amoxicillin/clavulanate, nitrofurantoin, isoniazid and ciprofloxacin; central nervous system (CNS) agents such as valproate, phenytoin, methyldopa, and lamotrigine; drugs used to treat endocrine disorders such as propylthiouracil, atorvastatin, and troglitazone; and amiodarone which is used to treat cardiovascular disorders. 32
On the other hand, pregnancy facilitates cholestasis and generates two pathologies accompanied by alterations of the liver profile. One of them is hyperemesis gravidarum which occurs during the first trimester of pregnancy. The other is intrahepatic cholestasis of pregnancy which appears in the second and third trimesters of pregnancy. 33 It manifests with pruritus and serum bile acid levels over 40 mmol/L and is associated with a high rate of fetal complications. Symptoms spontaneously resolve four to six weeks after delivery. 30,34
Intrahepatic cholestasis of pregnancy occurs in women who are heterozygous for MDR3 deficiency. Because MDR3 is a translocator in the canaliculi of the hepatocyte membrane, oral contraceptives have been counter indicated for these women. 35
Total parenteral nutrition has also been described as a cause of cholestasis. 26 Hemodynamic modifications have been identified within the acini and cholangioli of patients receiving this treatment. They empty into the hepatic artery rather than the portal vein. Similarly, the effects of prolonged fasting or digestive rest generate modifications in the enterohepatic circulation and changes in nutritional composition provided although these changes are not exactly the same as those that occur with enteral nutrition. 36,37
Sepsis has also been described as a cause of cholestasis in patients with Gram positive and Gram negative bacterial infections. Release of bacterial endotoxins such as lipopolysaccharides reduces recapture of bile by sodium-taurocholate receptors and by transporters that release bile salts.
This mechanism is mediated by proinflammatory cytokines such as interferon gamma and tumor necrosis factor. This facilitates lymphocytic infiltration into the ductal epithelium and reduces the secretion of bile in the bile ducts. 38,39 This phenomenon can manifest with hyperbilirubinemia and reach levels over 20 mg/L within the first 48 hours. It can also be increased by increased bilirubin loads (hemolysis, trauma or hematoma) and by liver alterations of bile flow (choledocholithiasis or intraductal inflammation). 40
Systemic disorders such as sarcoidosis initially generate pulmonary granulomatous and pulmonary nodule compromises. These appear in people between the ages of 20 and 40 and are accompanied by abdominal pain, nausea, vomiting, and high ALP and angiotensin converter enzyme levels. 39
Viral infections such as Epstein-Barr disease and cytomegalovirus can also cause systemic compromises related to cholestasis. Due to the nature of their DNA, these viruses compromise the liver. Cytomegalovirus, is capable of generating HIV cholangiopathy in patients with retrovirus infections although this is an unusual cause of sclerosing cholangitis. 3,39
In addition, immunoglobulin deposition diseases such as primary and secondary amyloidosis (due to plasma cell neoplasms such as macroglobulinemia or multiple myeloma) and secondary (due to systemic inflammatory diseases such as rheumatoid arthritis, tuberculosis, and osteomyelitis) are also thought to cause cholestasis. It is initially identified by an high ALP levels and hepatomegaly. Primary amyloidosis results from plasma cell neoplasms such as macroglobulinemia or from multiple myeloma while secondary amyloidosis is due to systemic inflammatory diseases such as rheumatoid arthritis, tuberculosis, and osteomyelitis. 29,39
Genetic disorders involving defects in the hepatocellular transport of bile are also reported to cause cholestasis. These disorders include Progressive Familial Intrahepatic Cholestasis (PFIC) Type 1 and Type 2 which are autosomal recessive disorders whose onset occurs during the neonatal period. Cholestasis appears in the first month of life, together with high ALP levels and normal gamma-glutamyl transferase levels in both PFIC 1 and PFIC 2.
PFIC 2 causes liver failure and progression to cirrhosis, hepatocellular carcinoma and cholangiocarcinoma. The onset of PFIC 3 occurs later n in childhood and is accompanied by high levels of gamma-glutamyl. 30,41
Another disorder, recurrent benign intrahepatic cholestasis is characterized by repeated episodes of pruritus and jaundice that last for weeks or months. Again, there are high levels of ALP subsequent to onset of pruritus, but the prognosis is benign. 42 Alagille syndrome is an autosomal dominant disorder which compromises multiple systems including the liver, heart, bone system, and eyes. It involves a mutation in the JAG1 gene and manifests itself with jaundice, hyperbilirubinemia, and high levels of ALP.
A less frequently occurring disorder, cystic fibrosis secondary to mutation of a gene involved with the chlorine-conducting transmembrane channels (CFTR) which modify anion transport and mucus clearance, results in respiratory compromise with bronchiectasis as well as liver damage and cirrhosis. 43
Like cystic fibrosis, Dubin-Johnson syndrome manifests itself through jaundice and hyperbilirubinemia (primarily of direct bilirubin,. The liver becomes black due to poor expression of the MRP2 transporter that alters the excretion of bile. 10,44
Diagnosis
The approach to cholestasis starts with a physical examination and the medical history. The physician must establish whether there are family precedents for cholestasis which might suggest PFIC and whether there is any surgical history of cholecystectomy which would probably indicate choledocholithiasis. 42. The medical history also should cover all drugs, medications, herbal remedies and alcohol consumption, any of which could cause this condition. 3,9,10
During the physical examination, the presence of painless jaundice, with or without a palpable mass in the right hypochondrium, may suggest malignant obstruction. Jaundice and abdominal pain together suggest choledocholithiasis. It can also manifest with cholangitis (fever, jaundice, pain in the right hypochondrium) due to partial or total obstruction of the bile duct. Intrahepatic cholestasis may appear with itching and fatigue.
The first step in studying cholestasis is measurement of ALP. Not specific to the liver, it can be found in the bones, kidneys, placenta, and white blood cells, but if high levels of ALP are found, and transaminase and/or bilirubin levels are not high, and there is no imaging evidence suggesting obstruction, measurement of gamma-glutamyl transferase and measurement of bone isoenzyme by electrophoresis or 5’-nucleotidase could be useful. It may also be appropriate to consider lifelong high levels of ALP and gender variance, since ALP levels are higher in women. 33,34
Similarly, blood tests for viral infections can exclude infectious hepatitis, and autoimmune etiologies should be ruled out by measuring ANA, ASMA, AMA, perinuclear ANCA, and immunoglobulin G and M levels. 45
Diagnostic images can be used to determine whether cholestasis is of intrahepatic or extrahepatic origin. Abdominal ultrasound is essential for excluding extrahepatic biliary obstruction, 10 but its use is technically difficult in obese people since it misses up to 60% of stones in the common bile duct of obese patients. 46
Other complementary studies of the bile duct include abdominal CT scans, although they do not adequately delineate the bile duct, 47 magnetic resonance cholangiography, endoscopic ultrasound, and endoscopic retrograde cholangiopancreatography. These tools are essential for identifying lesions that cause extrahepatic obstructions.
Abdominal ultrasound and magnetic resonance cholangiography can be used to characterize the intrahepatic bile duct, while endoscopic retrograde cholangiopancreatography contributes to instrumentation of the bile duct, extraction of stones in it and for taking biopsies when neoplastic lesions of the ducts are suspected. ERCP can help dilate bile duct stenoses and can be used to place stents in the common bile duct in cases of clinically manifest narrowness (Figure 3). 10
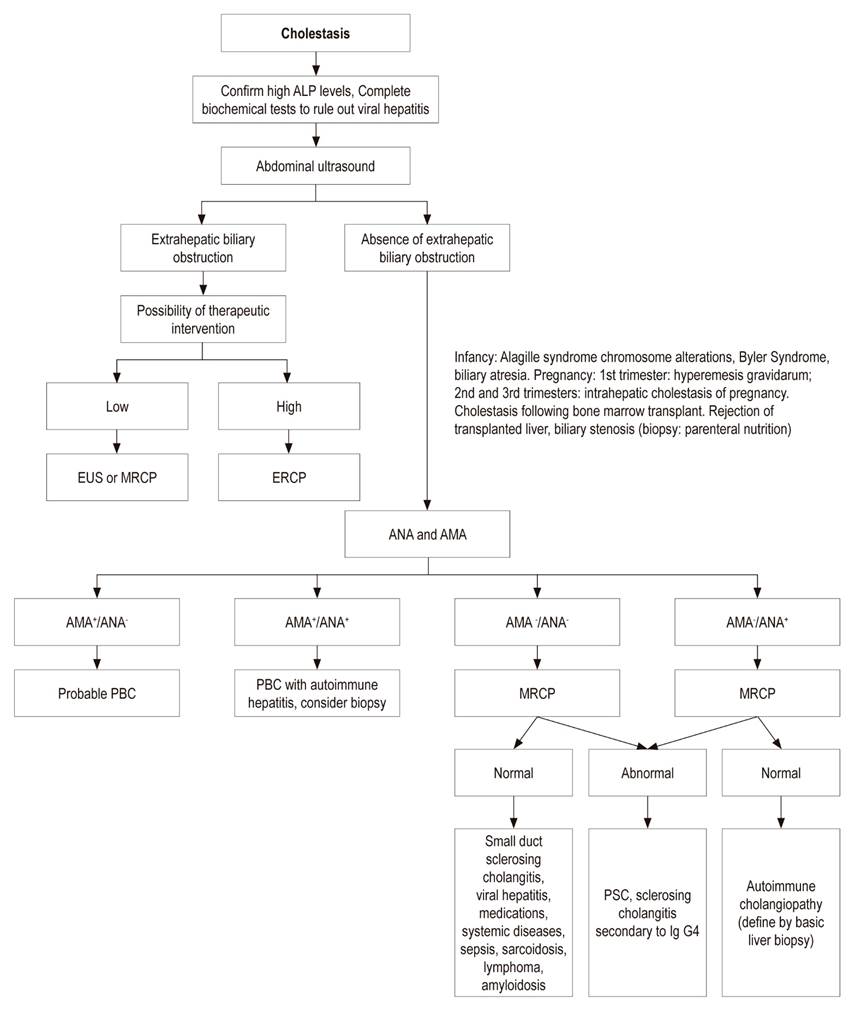
figure 3 cholestasis diagnostic algorithm. alp: alkaline phosphatase; eus: endoscopic ultrasound; mrcp: magnetic resonance cholangiography; ercp: endoscopic retrograde cholangiopancreatography; ana: antinuclear antibodies; ama: antimitochondrial antibodies; pbc: primary biliary cholangitis; ig g4: immunoglobulin g4. modified from reference 3. eus or mrcp; ercp endoscopic ultrasound or magnetic resonance cholangiography
Treatment
PBC and PSC are prototypes of chronic cholestatic diseases and are considered to be models of disorders for discussing medical management of cholestasis. 48 Treatment strategies seek to limit accumulation of bile acids and reduce bile acid reserves while trying to protect the liver by inducing choleresis (biliary excretion of acids) thus limiting damage to cholangiocytes and modulating inflammation caused by bile acids. 49
Management of PBC and PSC and other cholestatic diseases is based on the use of ursodeoxycholic acid which stimulates bile and bicarbonate flow in hepatocytes and cholangiocytes. It provides anti-apoptotic and anti-inflammatory effects 50 which can be used to identify response criteria after the start of treatment for PBC (Table 1). 51
Table 1 Criteria for responses after PBC treatment.
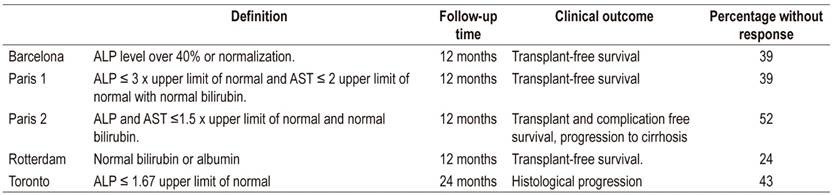
ALP: alkaline phosphatase; AST: aspartate aminotransferase. Modified from reference 51.
Nevertheless, there is insufficient evidence to demonstrate a survival benefit for PSC patients despite improvements in their liver profiles. 23 Other agents that are still in use are farnesoid X receptor (FXR) agonists like obeticholic acid, retinoid X receptor (RXR) agonists, pregnane X receptor (PXR) agonists, glucocorticoid receptor (GR) agonists and the peroxisome proliferator activated receptor (PPAR) agonists.
Fibrates such as fenofibrate and bezafibrate are transcriptional modifiers of bile formation which act on these receptors and prevent the accumulation of bile acids by reducing import and increasing export of these molecules in the hepatocytes. 49,52
For patients who do not respond to pharmacological measures, liver transplantation should be considered. It achieves 1 and 5-year survival rates of 83% and 78%, respectively. Nevertheless, the disease recurs in up to 8% of these patients by 5 years after and in 22% by 10 years after transplantation. 24
Conclusions
Cholestasis is a syndrome that compromises the synthesis, secretion and flow of bile and can lead to the risk of cirrhosis and portal hypertension. Early diagnosis and identification of subtle manifestations such as itching and changes in ALP levels are important, so clarity on hepatic origin and exclusion of other sources of ALP given its presence in other tissues are necessary.
Treatment seeks to decrease inflammation generated by bile acids and to modify bile acid reserves through excretion or blockade of enterohepatic circulation. Exclusion of complications such as osteoporosis and vitamin deficiency caused by this alteration, and prompt treatment of them when they do occur, are essential.
Acknowledgements
None declared by the authors
REFERENCES
1. McIntyre N. Cholestasis. En: Bircher J, Benhamou JP, McIntyre N, Rizzeto M, Rodés J. Oxford Textbook of Clinical Hepatology. Oxford: Oxford Medical Publications, 2a edición; 1999. p. 1574-9. [ Links ]
2. Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol. 2007;5(7):776-82. https://doi.org/10.1016/j.cgh.2007.05.008 [ Links ]
3. Pérez Fernández T, López Serrano P, Tomás E, Gutiérrez ML, Lledó JL, Cacho G, et al. Diagnostic and therapeutic approach to cholestatic liver disease. Rev Esp Enferm Dig. 2004;96(1):60-73. https://doi.org/10.4321/S1130-01082004000100008 [ Links ]
4. Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339(17):1217-27. https://doi.org/10.1056/NEJM199810223391707 [ Links ]
5. Zollner G, Trauner M. Mechanisms of cholestasis. Clin Liver Dis. 2008;12(1):1-26, vii. https://doi.org/10.1016/j.cld.2007.11.010 [ Links ]
6. Elferink RO. Cholestasis. Gut. 2003;52 Suppl 2(Suppl 2):ii42-ii48. https://doi.org/10.1136/gut.52.suppl_2.ii42 [ Links ]
7. Meier PJ. Molecular mechanisms of hepatic bile salt transport from sinusoidal blood into bile. Am J Physiol. 1995;269(6 Pt 1):G801-12. https://doi.org/10.1152/ajpgi.1995.269.6.G801 [ Links ]
8. Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65(16):2461-83. https://doi.org/10.1007/s00018-008-7568-6 [ Links ]
9. Maillette de Buy Wenniger L, Beuers U. Bile salts and cholestasis. Dig Liver Dis. 2010;42(6):409-18. https://doi.org/10.1016/j.dld.2010.03.015 [ Links ]
10. Siddique A, Kowdley KV. Approach to a patient with elevated serum alkaline phosphatase. Clin Liver Dis. 2012;16(2):199-229. https://doi.org/10.1016/j.cld.2012.03.012 [ Links ]
11. Assis DN. Chronic Complications of Cholestasis: Evaluation and Management. Clin Liver Dis. 2018;22(3):533-544. https://doi.org/10.1016/j.cld.2018.03.014 [ Links ]
12. Shen YM, Wu JF, Hsu HY, Ni YH, Chang MH, Liu YW, et al. Oral absorbable fat-soluble vitamin formulation in pediatric patients with cholestasis. J Pediatr Gastroenterol Nutr. 2012;55(5):587-91. https://doi.org/10.1097/MPG.0b013e31825c9732 [ Links ]
13. Shneider BL, Magee JC, Bezerra JA, Harber B, Karpen SJ, Raghunathan T, et al. Efficacy of fat-soluble vitamin supplementation in infants with biliary atresia. Pediatrics. 2012;130(3):e607-e614. https://doi.org/10.1097/MPG.0b013e31825c9732 [ Links ]
14. Craddock AL, Love MW, Daniel RW, et al. Expression and transport properties of the human ileal and renal sodium- dependent bile acid transporter. Am J Physiol 1998; 274 (1): 157-69. https://doi.org/10.1152/ajpgi.1998.274.1.G157 [ Links ]
15. Kwo PY, Cohen SM, Lim JK. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Am J Gastroenterol. 2017;112(1):18-35. https://doi.org/10.1038/ajg.2016.517 [ Links ]
16. Schiele F, Henny J, Hitz J, Petitclerc C, Gueguen R, Siest G. Total bone and liver alkaline phosphatases in plasma: biological variations and reference limits. Clin Chem. 1983;29(4):634-41. https://doi.org/10.1093/clinchem/29.4.634 [ Links ]
17. Stigbrand T. Present status and future trends of human alkaline phosphatases. Prog Clin Biol Res. 1984;166:3-14. [ Links ]
18. Sharma U, Pal D, Prasad R. Alkaline phosphatase: an overview. Indian J Clin Biochem. 2014;29(3):269-278. https://doi.org/10.1007/s12291-013-0408-y [ Links ]
19. Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci. 1985;30(3):211-4. https://doi.org/10.1007/BF01347885 [ Links ]
20. Lee DH, Silventoinen K, Hu G, Jacobs DR, Jousilahti P, Sundvall J, et al. Serum gamma-glutamyltransferase predicts non-fatal myocardial infarction and fatal coronary heart disease among 28,838 middle-aged men and women. Eur Heart J. 2006;27(18):2170-6. https://doi.org/10.1093/eurheartj/ehl086 [ Links ]
21. Kim HW, Lee SH, Lee DH. Relationship of serum gamma-glutamyltransferase levels with pulmonary function and chronic obstructive pulmonary disease. Lung. 2014;192(5):719-27. https://doi.org/10.1007/s00408-014-9616-3 [ Links ]
22. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145-172. https://doi.org/10.1016/j.jhep.2017.03.022 [ Links ]
23. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237-67. https://doi.org/10.1016/j.jhep.2009.04.009 [ Links ]
24. Patel A, Seetharam A. Primary Biliary Cholangitis: Disease Pathogenesis and Implications for Established and Novel Therapeutics. J Clin Exp Hepatol. 2016;6(4):311-318. https://doi.org/10.1016/j.jceh.2016.10.001 [ Links ]
25. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, et al. Primary biliary cirrhosis. Hepatology. 2009;50(1):291-308. https://doi.org/10.1002/hep.22906 [ Links ]
26. Jansen PL, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology. 2017;65(2):722-738. https://doi.org/10.1002/hep.28965 [ Links ]
27. Williamson KD, Chapman RW. New Therapeutic Strategies for Primary Sclerosing Cholangitis. Semin Liver Dis. 2016;36(1):5-14. https://doi.org/10.1055/s-0035-1571274 [ Links ]
28. Lindor KD, Kowdley KV, Harrison ME; American College of Gastroenterology. ACG Clinical Guideline: Primary Sclerosing Cholangitis. Am J Gastroenterol. 2015;110(5):646-59. https://doi.org/10.1038/ajg.2015.112 [ Links ]
29. Smit WL, Culver EL, Chapman RW. New Thoughts on Immunoglobulin G4-Related Sclerosing Cholangitis. Clin Liver Dis. 2016;20(1):47-65. https://doi.org/10.1016/j.cld.2015.08.004 [ Links ]
30. Nguyen KD, Sundaram V, Ayoub WS. Atypical causes of cholestasis. World J Gastroenterol. 2014;20(28):9418-26. http://dx.doi.org/10.3748/wjg.v20.i28.9418 [ Links ]
31. Bhamidimarri KR, Schiff E. Drug-induced cholestasis. Clin Liver Dis. 2013;17(4):519-31, vii. https://doi.org/10.1016/j.cld.2013.07.015 [ Links ]
32. Padda MS, Sanchez M, Akhtar AJ, Boyer JL. Drug-induced cholestasis. Hepatology . 2011;53(4):1377-1387. https://doi.org/10.1002/hep.24229 [ Links ]
33. Floreani A, Gervasi MT. New Insights on Intrahepatic Cholestasis of Pregnancy. Clin Liver Dis. 2016;20(1):177-89. https://doi.org/10.1016/j.cld.2015.08.010 [ Links ]
34. Lammert F, Marschall HU, Glantz A, Matern S. Intrahepatic cholestasis of pregnancy: molecular pathogenesis, diagnosis and management. J Hepatol. 2000;33(6):1012-21. https://doi.org/10.1016/S0168-8278(00)80139-7 [ Links ]
35. Jacquemin E, De Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120(6):1448-58. https://doi.org/10.1053/gast.2001.23984 [ Links ]
36. Guglielmi FW, Regano N, Mazzuoli S, Fregnan S, Leogrande G, Guglielmi A, et al. Cholestasis induced by total parenteral nutrition. Clin Liver Dis. 2008;12(1):97-110, viii. https://doi.org/10.1016/j.cld.2007.11.004 [ Links ]
37. Lauriti G, Zani A, Aufieri R, Cananzi M, Chiesa PL, Eaton S, et al. Incidence, prevention, and treatment of parenteral nutrition-associated cholestasis and intestinal failure-associated liver disease in infants and children: a systematic review. JPEN J Parenter Enteral Nutr. 2014;38(1):70-85. https://doi.org/10.1177/0148607113496280 [ Links ]
38. Geier A, Fickert P, Trauner M. Mechanisms of disease: mechanisms and clinical implications of cholestasis in sepsis. Nat Clin Pract Gastroenterol Hepatol. 2006;3(10):574-85. https://doi.org/10.1038/ncpgasthep0602 [ Links ]
39. Delemos AS, Friedman LS. Systemic causes of cholestasis. Clin Liver Dis. 2013;17(2):301-17. https://doi.org/10.1016/j.cld.2012.11.001 [ Links ]
40. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14(1):55-66. https://doi.org/10.1038/nrgastro.2016.168 [ Links ]
41. Davit-Spraul A, Gonzáles E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 2010;30(2):134-46. https://doi.org/10.1055/s-0030-1253223 [ Links ]
42. Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. 2004;8(1):133-49, vii. https://doi.org/10.1016/S1089-3261(03)00133-8 [ Links ]
43. Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519-2531. https://doi.org/10.1016/S0140-6736(16)00576-6 [ Links ]
44. Memon N, Weinberger BI, Hegyi T, Aleksunes LM. Inherited disorders of bilirubin clearance. Pediatr Res. 2016;79(3):378-86. https://doi.org/10.1038/pr.2015.247 [ Links ]
45. Marzorati S, Invernizzi P, Lleo A. Making Sense of Autoantibodies in Cholestatic Liver Diseases. Clin Liver Dis. 2016;20(1):33-46. https://doi.org/10.1016/j.cld.2015.08.003 [ Links ]
46. Shea JA, Berlin JA, Escarce JJ, Clarke JR, Kinosian BP, Cabana MD, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med. 1994;154(22):2573-81. https://doi.org/10.1001/archinte.1994.00420220069008 [ Links ]
47. Balci NC, Befeler AS, Leiva P, Pilgram TK, Havlioglu N. Imaging of liver disease: comparison between quadruple-phase multidetector computed tomography and magnetic resonance imaging. J Gastroenterol Hepatol. 2008;23(10):1520-7. https://doi.org/10.1111/j.1440-1746.2008.05434.x [ Links ]
48. Samant H, Manatsathit W, Dies D, Shokouh-Amiri H, Zibari G, Boktor M, et al. Cholestatic liver diseases: An era of emerging therapies. World J Clin Cases. 2019;7(13):1571-1581. https://doi.org/10.12998/wjcc.v7.i13.1571 [ Links ]
49. Arab JP, Cabrera D, Arrese M. Bile Acids in Cholestasis and its Treatment. Ann Hepatol. 2017;16(Suppl. 1: s3-105.):s53-s57. https://doi.org/10.5604/01.3001.0010.5497 [ Links ]
50. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol. 2015;62(1 Suppl):S25-37. https://doi.org/10.1016/j.jhep.2015.02.023 [ Links ]
51. Chazouillères O. Novel Aspects in the Management of Cholestatic Liver Diseases. Dig Dis. 2016;34(4):340-6. https://doi.org/10.1159/000444544 [ Links ]
52. Czul F, Levy C. Novel Therapies on Primary Biliary Cirrhosis. Clin Liver Dis. 2016;20(1):113-30. https://doi.org/10.1016/j.cld.2015.08.006 [ Links ]
Received: March 16, 2019; Accepted: December 18, 2019
 Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons
