











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El término colestasis proviene de las palabras griegas chole, que significa bilis, y stasis, que traduce quieto. Esta definición involucra el síndrome generado por la alteración en la síntesis, la secreción o el flujo de la bilis, a través del tracto biliar. Se manifiesta a partir de la elevación de forma inicial de los niveles de la fosfatasa alcalina (Alkaline phosphatase, ALP) sérica y la gamma-glutamil transferasa 1, así como con la presencia de prurito generalizado sin lesiones en la piel y fatiga. Aunque la ictericia es un signo importante en la colestasis, puede estar ausente, particularmente en adultos con enfermedades crónicas colestásicas que pueden ser asintomáticos 2.
Esta entidad se ha clasificado en el segmento involucrado en la excreción de la bilis, desde la etiología intrahepática, en la cual se evidencia un compromiso en el citoplasma hepatocelular y en los conductos biliares de tamaño mediano (hasta 400 µm de diámetro), y las causas extrahepáticas, que comprometen los conductos biliares de gran tamaño. En consecuencia, las causas que generan la obstrucción de la vía biliar son producidas por cálculos, tumores pancreáticos o biliares, así como por metástasis hiliares 3.
Fisiopatología
Para comprender el origen de la colestasis, se debe partir desde la composición del lobulillo hepático. Esta unidad surge de la porción funcional más pequeña en el hígado, donde los hepatocitos se organizan en placas a lo largo del flujo sanguíneo, desde la vena porta hasta la vena central. Dentro de estas placas, los hepatocitos forman una luz tubular llamada el canalículo, en la cual se da la formación inicial de la bilis.
Estos hepatocitos contienen dos sistemas de toma y exportación, localizados en la porción basolateral (sinu-soidal) y canalicular (apical) de la membrana de los hepatocitos y los colangiocitos, acompañados por un proceso osmótico que permite la generación de un flujo de agua que produce la secreción de bilis, desde los conductos biliares más pequeños hasta los más grandes 4,5,6 (Figura 1).

Figura 1 Sistemas de transporte de ácidos biliares. NTCP: péptido cotransportador de sodio-taurocolato (Na-Taurocholate Cotransporting Polypeptide); OATP2: polipéptido transportador de aniones orgánicos 2 (Organic Anion Transporting Polypeptide); BSEP: bomba exportadora de sales biliares (Bile Salt Export Pump); MRP2: proteína de resistencia a múltiples fármacos tipo 2 (Multidrug Resistance-associated Protein 2); MDR3: resistencia a múltiples fármacos tipo 3 (Multidrug Resistance 3); MDR1: resistencia a múltiples fármacos tipo 1 (Multidrug Resistance 1); OST a/b: transportador orgánico de solutos a/b (Organic Solute Transporter alpha and beta); MRP3: proteína de resistencia a múltiples fármacos tipo 3 (Multidrug Resistance-associated Protein 2); MRP4: proteína de resistencia a múltiples fármacos tipo 4 (Multidrug Resistance-associated Protein 4); CFTR: regulador transmembrana de la fibrosis quística (Cystic Fibrosis Transmembrane conductance Regulator); ASBT: transportador apical de sales biliares dependiente de sodio (Apical Sodium-Bile acid Transporter); AB: ácido biliar; OA: anión orgánico (Organic Anion); PC: fosfatidilcolina (Phosphatidylcholine). Tomada de la referencia 5.
Los ácidos biliares captados ingresan por medio de receptores del péptido cotransportador de sodio-taurocolato (Na-Taurocholate Cotransporting Polypeptide, NTCP), los cuales permiten concentrar las sales biliares en el hepatocito 6,7,8, así como otros polipéptidos transportadores de aniones orgánicos (Organic Anion Transporting Polypeptide, OATP2/OATP1B1), localizados en las membranas basolaterales de los hepatocitos 5.
Los ácidos biliares son excretados en la bilis por la bomba exportadora de ácidos biliares (Bile Salt Export Pump, BSEP) canaliculares y por la proteína exportadora canalicular conjugada MRP2 (Multidrug Resistance-associated Protein 2).
El transportador MRP2 (ABCC2) secreta aniones orgánicos como la bilirrubina conjugada, el glutatión e incluso antibióticos como la ceftriaxona 6. Otros transportadores localizados en la membrana canalicular son el MDR1 (Multidrug Resistance 1), la glicoproteína P (ABCB1) -que bombea componentes orgánicos y medicamentos catiónicos o con carga neutra fuera de la célula- y el MDR3 (Multidrug Resistance 3), el cual excreta fosfolípidos como la fosfatidilcolina que, en combinación con el colesterol y los ácidos biliares, forma las micelas 4,5,6 (Figura 1).
La bilis que se ha formado con las sales biliares primarias (colato y quenodesoxicólico), por la modificación enzimática del colesterol, confiere características más hidrofílicas y conforma las micelas, cuya función primordial es la toma de lípidos del intestino 8,9. Sin embargo, son potencialmente peligrosas para la integridad de la membrana celular en condiciones de colestasis 6.
Entre tanto, los colangiocitos permiten la formación de la vía biliar y proveen otra serie de receptores que facilitan la reabsorción de ácidos biliares como el transportador apical de ácidos biliares dependiente de sodio (Apical Sodium-Bile acid Transporter, ASBT) y el canal de cloro CFTR (Cystic Fibrosis Transmembrane conductance Regulator), que es el regulador transmembrana de fibrosis quística 5.
Una vez las sales biliares se han secretado en el lumen intestinal, se recapturan por circulación enterohepática en el íleon, mediante un ASBT 9, hasta llegar al hepatocito, donde son nuevamente tomadas por el receptor NTCP, el cual permite concentrar las sales biliares en el hepatocito 5,6. Asimismo, en la membrana basolateral del hepatocito se localizan otros receptores denominados MRP3 (Multidrug Resistance-associated Protein 3) y MRP4 (Multidrug Resistance-associated Protein 4), así como el transportador orgánico de solutos (Organic Solute Transporter alpha and beta, OST a/b). Estos son una ruta de excreción alternativa para los ácidos biliares y otros aniones orgánicos y se dirigen hacia la circulación sistémica 5.
La colestasis resulta de un defecto funcional en la formación de la bilis en el hepatocito, o de una alteración en la secreción y el flujo de esta a nivel de los conductos biliares 4,5 (Figura 2).
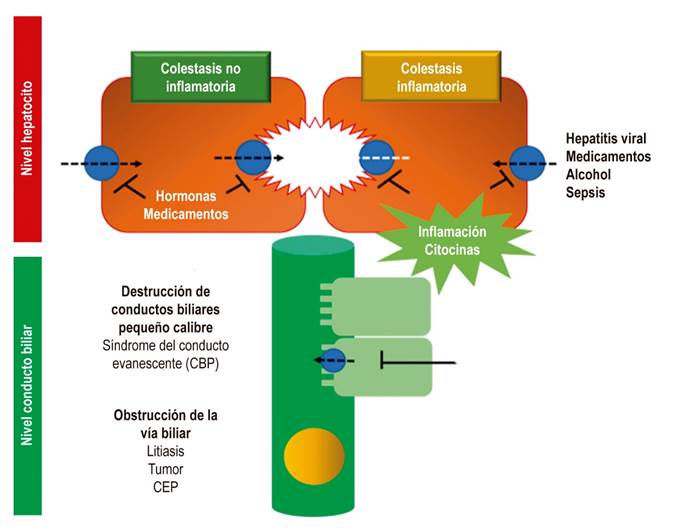
Manifestaciones clínicas
La colestasis puede tener un espectro de síntomas generados por la acumulación de sustancias en el hígado y en la sangre, que generalmente son excretados en la bilis. Los pacientes con este síndrome pueden encontrarse asintomáticos e incluso referir prurito e ictericia. Sin embargo, la ictericia puede estar ausente en adultos con enfermedades hepáticas colestásicas crónicas 2.
Un hallazgo importante es la esteatorrea, que se define como la pérdida >10 g de grasa fecal por día, luego de una ingesta de 70 g/d (10). Este fenómeno es secundario a una concentración inadecuada de bilis posprandial en el intestino delgado, lo que genera una malabsorción de grasas y vitaminas liposolubles que usualmente ayudan en la absorción de estos elementos 3,9. La esteatorrea puede acompañarse de pérdida de peso, así como de la presencia de acropaquía o dedos hipocráticos 2.
Este déficit de vitaminas liposolubles tiene un amplio rango de síntomas neurológicos como ceguera nocturna (deficiencia de vitamina A); hiporreflexia o ataxia, como síntomas secundarios a una mielopatía (déficit de vitamina E); coagulopatía (deficiencia de vitamina K), y la presencia de trastornos del sistema osteomuscular con osteomalacia, osteoporosis y fracturas (deficiencia de vitamina D y calcio) 3,9,10. Algunos estudios han sugerido que la hiperbilirrubinemia persistente >2-3 mg/dL se asocia con la deficiencia de vitaminas liposolubles 11,12,13.
Además, se ha documentado la asociación de la osteoporosis y la deficiencia de vitaminas liposolubles con la prolongación del tiempo de protrombina, dado que los factores 2, 7, 9 y 10 son dependientes de la vitamina K, la cual no puede ser absorbida durante los episodios de colestasis 14.
Asimismo, algunas enfermedades asociadas con la colestasis, como la colangitis biliar primaria, pueden continuar su progresión hasta presentar manifestaciones de hipertensión portal, dadas por ascitis, encefalopatía y hemorragia de las vías digestivas altas 10. Así, durante el examen físico es posible hallar xantomas (depósitos de colesterol en los tendones o prominencias óseas en los codos y las rodillas) o xantelasmas (depósitos de lípidos en los pliegues periorbitarios) 2,11.
Alteraciones paraclínicas
El examen más sensible para la identificación de la colestasis es la medición de ácidos biliares séricos. Sin embargo, este estudio no está disponible con frecuencia. Por tanto, es necesario acudir a los marcadores bioquímicos de colestasis crónica, que son la ALP y la gamma-glutamil transferasa 2.
La fosfatasa puede elevarse por causas fisiológicas asociadas a un ascenso de dicha enzima. Esta elevación ocurre en los 3 primeros meses de vida y la pubertad, y se incrementa de forma gradual entre los 40 y 65 años, especialmente en mujeres y durante el embarazo 2,10. Mientras tanto, los adolescentes hombres pueden alcanzar entre 2 y 5 veces el valor normal de los adultos, lo que se correlaciona con el crecimiento óseo 15.
Otras variaciones fisiológicas pueden encontrarse en los afroamericanos, que tienen entre un 10 y un 15 % más de niveles séricos de ALP, así como en los fumadores, que pueden alcanzar hasta un 10 % más, en comparación con las personas no fumadoras 10. De igual forma, las personas con un grupo sanguíneo O y B pueden presentar una elevación de la ALP, luego de una comida con alto contenido de grasa, a raíz del influjo intestinal de la misma enzima 2,10,11.
Las causas patológicas que generan un incremento de la ALP varían desde enfermedades óseas como fracturas, enfermedad de Paget, osteomalacia, deficiencia de vitamina D, falla cardíaca o renal, hipertiroidismo e hiperparatiroidismo, hasta neoplasias hematológicas como leucemia y linfoma, o de células renales 10.
Este fenómeno surge tanto por la presencia de isoformas específicas en el tejido, como las placentarias (placental-like alkaline phosphatase, PLALP, o la isoenzima Regan), las intestinales (Intestinal Alkaline Phosphatase, IALP), las de hígado/hueso/renal (Liver/Bone/Kidney, ALP) y las células germinales de ALP (Germ Cell Alkaline Phosphatase, GCALP, o la isoenzima NAGAO), así como por su localización en la superficie celular, lo cual hidroliza los ésteres de monofosfato a pH alto, con la liberación de fosfato inorgánico 16,17,18.
Ante la presencia de una ALP elevada, se debe considerar si este incremento tiene una fuente intrahepática o extrahepática (principalmente en los huesos, el intestino, los leucocitos y la placenta). Por tanto, se requiere la toma de gamma-glutamil transferasa o una medición de las isoenzimas de ALP que considere un origen óseo 2,11. Esta actividad ósea cobija alrededor de la mitad de la ALP en los adultos, por lo que esta es la isoenzima más importante y útil para su estudio 18.
No obstante, la gamma-glutamil transferasa presenta una baja especificidad para enfermedades colestásicas, dado que puede elevarse hasta en un 50 % de los pacientes alcohólicos sin evidencia de enfermedad hepática 19. Esto también puede ocurrir en otras etiologías entre las que se incluyen enfermedades pancreáticas, infarto de miocardio 20, falla renal y enfisema 21, y en pacientes que consumen medicamentos como la fenitoína y los barbitúricos.
En consecuencia, este no es un examen que deba ser usado para la tamización de enfermedades hepáticas subyacentes, en ausencia de anormalidad en las otras pruebas hepáticas 15. La bilirrubina sérica, en su forma conjugada, puede estar elevada (o no) en presencia de una colestasis 2,22,23.
Etiología
Dentro de las múltiples causas de colestasis pueden identificarse alteraciones genéticas que aparecen en la infancia y trastornos autoinmunes, sistémicos o secundarios a medicamentos. Sin embargo, una forma de categorizar la etiología es dividirla en causas intrahepáticas y extrahepáticas 3.
Las causas intrahepáticas son ocasionadas, generalmente, por condiciones inflamatorias y destructivas que suelen denominarse como el síndrome del conducto evanescente 3,5. La colangitis biliar primaria (CBP), anteriormente conocida como cirrosis biliar primaria, es una de las causas más frecuentes de colestasis de origen autoinmune y genera una colangitis granulomatosa linfocítica que involucra a los conductos biliares de pequeño calibre 3,24,25.
Por lo general, la CBP se presenta en mujeres (relación 9:1) entre los 40 y 60 años; se acompaña de anticuerpos antimitocondriales (Anti-mitochondrial antibodies, AMA) positivos (títulos >1:40) dirigidos contra la subunidad E2 de la piruvato deshidrogenasa 22. Su diagnóstico se realiza a partir de 2 de 3 criterios: la presencia de colestasis por elevación de la ALP, los AMA positivos y la evidencia histopatológica de colangitis no supurativa con destrucción de los conductos biliares interlobulares de mediano y pequeño calibre 26.
Entre tanto, la colangitis esclerosante primaria (CEP) es una entidad que predomina en los hombres 2:1 con respecto a las mujeres y tiene una edad media de aparición de, aproximadamente, 40 años. Esta afecta los conductos biliares intrahepáticos y extrahepáticos, lo que genera estrecheces, con posterior fibrosis y cirrosis 27,28. El diagnóstico de esta enfermedad involucra una combinación de criterios por imágenes que pueden mostrar estenosis localizadas a lo largo de todo el árbol biliar, las cuales son identificables mediante una colangioresonancia o una colangiopancreatografía retrógrada endoscópica. Los cambios en el conducto biliar pueden observarse a partir de la biopsia hepática, en la que se evidencia una fibrosis concéntrica periductal o en «piel de cebolla» 27.
Asimismo, se pueden identificar anticuerpos antimúsculo liso (anti-smooth muscle antibody, ASMA), anticuerpos antinucleares (AntiNuclear Antibodies, ANA) y anticuerpos contra el citoplasma de neutrófilos (Antineutrophil Cytoplasmic Antibodies, ANCA) hasta en un 50 % de los pacientes; sin embargo, estos no son específicos de dicha entidad 28.
Otras enfermedades con sustrato autoinmune, que pueden generar colestasis y que comparten la misma fisiopatología de la fibrosis periductal y la pérdida progresiva de los conductos biliares, son la colangiopatía por inmuno-globulina G4; el rechazo crónico ductopénico en trasplante hepático y la enfermedad de injerto contra huésped, la cual aparece en los primeros 100 días luego del trasplante alogénico de células hematopoyéticas.
Incluso el linfoma (tanto Hodgkin como no Hodgkin) podría manifestarse hasta en un 10 % de los pacientes con ictericia y ductopenia progresiva (síndrome de conducto evanescente) y en un 40 % de aquellos con niveles elevados de ALP. Se acompaña de los síntomas clásicos B como fiebre y pérdida de peso, y presenta un espectro que abarca desde una lesión solitaria hasta el compromiso infiltrativo difuso, lo que responde tanto a la quimioterapia como a la radioterapia 3,29.
El uso de medicamentos puede generar lesiones hepáticas (enfermedad hepática inducida por fármacos [Drug Induced Liver Injury, DILI]) y desembocar en una falla hepática fulminante (13 % de los pacientes en Estados Unidos). Estas lesiones se manifiestan con un patrón hepatocelular, y una elevación de transaminasas, o un patrón colestásico con elevación de la ALP >2 veces el límite superior de la normalidad 30.
Así, los cambios generados por los medicamentos pueden alcanzar desde una discreta inflamación del parénquima y ductopenia leve hasta una inflamación progresiva, fibrosis y la pérdida de los conductos biliares 31. Entre los principales fármacos identificados como desencadenantes de este fenómeno, se encuentran los antibióticos (amoxicilina/clavulanato, nitrofurantoína, isoniazida y ciprofloxacina, entre otros), agentes del sistema nervioso central (SNC) (valproato, fenitoína, metildopa, lamotrigina) y medicamentos usados en los trastornos endocrinos (propiltiouracilo, atorvastatina, troglitazona) y cardiovasculares (amiodarona) 32.
Por otra parte, el embarazo facilita la presencia de colestasis y genera dos patologías acompañadas de alteraciones en el perfil hepático. Una de ellas es la hiperémesis gravídica, que ocurre durante el primer trimestre del embarazo, y otra es la colestasis intrahepática del embarazo, que aparece en el segundo y el tercer trimestre de la gestación 33. Esta última se manifiesta con prurito, una elevación de los ácidos biliares séricos (niveles >40 mmol/L se asocian con una mayor tasa de complicaciones fetales) y la resolución espontánea de los síntomas entre las semanas 4 y 6 posteriores al parto 30,34.
De igual forma, existe una particularidad en las mujeres que consumen anticonceptivos orales, dado que este síndrome se presenta en mujeres heterocigotas para la deficiencia de MDR3, el cual es un translocador en el canalículo de la membrana del hepatocito 35.
La nutrición parenteral total también se ha descrito como una causa de colestasis 26. En efecto, en los pacientes que reciben este tratamiento se han identificado modificaciones hemodinámicas dentro del acino y los colangiolos, desde la arteria hepática, en lugar de ser recibidos por la vena porta. Asimismo, los efectos del ayuno prolongado o el reposo digestivo generan modificaciones en la circulación enterohepática y cambios en la composición nutricional aportada, que no será exactamente igual a la nutrición enteral 36,37.
La sepsis también se ha descrito como una causa de colestasis que involucra a los pacientes con infecciones bacterianas, tanto por gérmenes Gram positivos como Gram negativos, mediante la liberación de endotoxinas bacterianas, como los lipopolisacáridos, que reducen la recaptura de la bilis en los receptores de sodio-taurocolato y en los transportadores que liberan las sales biliares.
Dicho mecanismo está mediado por la presencia de citocinas proinflamatorias como el interferón gamma y el factor de necrosis tumoral, lo cual facilita la infiltración linfocítica en el epitelio ductal y reduce la secreción de bilis en los conductos biliares 38,39. Este fenómeno puede manifestarse con hiperbilirrubinemia y alcanzar niveles >20 mg/L dentro de las primeras 48 h, así como también puede incrementarse por cargas aumentadas de bilirrubina (hemólisis trauma o hematoma) y por alteraciones hepáticas en el flujo biliar (coledocolitiasis o inflamación intraductal) 40.
Los trastornos sistémicos como la sarcoidosis, que inicialmente genera un compromiso granulomatoso pulmonar, y los nódulos pulmonares, que aparecen en personas entre los 20 y 40 años, se acompañan de dolor abdominal, náuseas, vómito, una elevación de la ALP y de la enzima convertidora de angiotensina 39.
Otras causas del compromiso sistémico con colestasis son las infecciones por virus, como el Epstein-Barr y el citomegalovirus. Por la naturaleza de su ADN, estos virus comprometen el hígado y, en el caso del citomegalovirus, su presencia es capaz de generar una colangiopatía por VIH en pacientes con infección por retrovirus. Esta constituye una causa inusual de colangitis esclerosante 3,39.
De forma simultánea, las enfermedades de depósito de inmunoglobulinas, como la amiloidosis primaria (por neoplasias de células plasmáticas como la macroglobulinemia o el mieloma múltiple) y secundaria (por enfermedades inflamatorias sistémicas como la artritis reumatoide, la tuberculosis y la osteomielitis) también se han considerado como causantes de colestasis -la cual es inicialmente identificada por un aumento de la ALP- y de hepatomegalia 29,39.
Los trastornos genéticos que involucran defectos en el transporte hepatocelular de la bilis han sido descritos como causantes de colestasis. Entre estos trastornos se incluye la colestasis intrahepática progresiva familiar (Progressive Familial Intrahepatic Cholestasis, PFIC), la cual hace parte de los trastornos autosómicos recesivos de aparición desde el período neonatal. Dicha alteración se presenta con colestasis de aparición en el primer mes de vida, junto con una ALP elevada y una gamma-glutamil transferasa normal, tanto en la PFIC 1 como en la PFIC 2.
Esta última causa una falla hepática con progresión a cirrosis hepatocarcinoma y colangiocarcinoma, mientras que la PFIC 3 tiene una presentación más tardía en la infancia, acompañada de una gamma-glutamil elevada 30,41.
Entre tanto, la colestasis intrahepática benigna recurrente se caracteriza por episodios repetidos de prurito e ictericia, que duran semanas o meses, y están acompañados de una ALP elevada, posterior al inicio de estos episodios de prurito con un pronóstico benigno 42. Por su parte, el síndrome de Alagille es un trastorno autosómico dominante, con compromiso multisistémico (hígado, corazón, sistema óseo y ojos), que involucra una mutación en el gen JAG1 y se manifiesta con ictericia, hiperbilirrubinemia y una ALP elevada.
Otra entidad menos frecuente es la fibrosis quística secundaria a la mutación del gen que involucra a los canales transmembrana conductores de cloro (CFTR), los cuales modifican el trasporte aniónico, así como la depuración del moco, y presentan un compromiso respiratorio con bronquiectasias y en el hígado, con cirrosis 43.
Además de la fibrosis quística, el síndrome de Dubin-Johnson se manifiesta mediante la ictericia y la hiperbilirrubinemia predominantemente directa, y con cambios en la coloración del hígado, la cual se torna de un aspecto negro, por causa de una deficiente expresión del transportador MRP2 que altera la excreción de la bilis 10,44.
Diagnóstico
El abordaje de la colestasis parte de la historia clínica y del examen físico, con los cuales se indican los antecedentes médicos y se establece si existen precedentes familiares de colestasis (lo que sugiere una colestasis intrahepática familiar) 42, antecedentes quirúrgicos de colecistectomía (probable coledocolitiasis) e incluso se indaga sobre las terapias farmacológicas usadas y el consumo de remedios herbales y de alcohol, que podrían ser causas de esta afección 3,9,10.
Durante el examen físico, la presencia de ictericia indolora, con o sin masa palpable en el hipocondrio derecho, puede sugerir una obstrucción maligna, mientras que la coledocolitiasis puede presentarse con ictericia y dolor abdominal. También se puede manifestar con colangitis (fiebre, ictericia, dolor en el hipocondrio derecho), por causa de una obstrucción parcial o total de la vía biliar. Entre tanto, la colestasis intrahepática puede aparecer junto con prurito y fatiga.
Así, el primer paso para estudiar la colestasis es la medición de la ALP, la cual no es específica del hígado, sino que puede encontrarse en los huesos, el riñón, la placenta y en los glóbulos blancos. En caso de hallarse una elevación de la ALP sin un aumento de transaminasas o hiperbilirrubinemia, y sin evidencias imagenológicas que sugieran la existencia de un componente obstructivo, sería útil la medición de la isoenzima ósea por electroforesis o 5’-nucleotidasa y gamma-glutamil transferasa. También puede resultar oportuno considerar las elevaciones mismas de la ALP en el transcurso de la vida y la variación según el género, dado que esta es más elevada en las mujeres 33,34.
Del mismo modo, se pueden complementar los estudios a partir de la toma de serologías virales, a fin de excluir el diagnóstico de hepatitis de etiología infecciosa. Las etiologías autoinmunes deben descartarse por medio de la medición de los ANA, los ASMA, los AMA, los ANCA de fracción perinuclear y los niveles de inmunoglobulina G y M 45.
De forma simultánea, las imágenes diagnósticas permiten orientar la colestasis de origen intrahepático o extrahepático. La ultrasonografía abdominal es fundamental para excluir la presencia de una obstrucción biliar extrahepática 10. Sin embargo, su uso tiene dificultades técnicas en personas obesas, dado que omite hasta el 60 % de los cálculos en el colédoco de este tipo de pacientes 46.
Otros estudios complementarios en la vía biliar son la tomografía de abdomen -aunque esta no delinea de forma adecuada la vía biliar- 47, la colangioresonancia magnética, la ultrasonografía endoscópica o ecoendoscopia y la colangiopancreatografía retrógrada endoscópica. Estas herramientas son fundamentales para la identificación de las lesiones causantes de la obstrucción extrahepática.
Las dos primeras pruebas permiten una caracterización de la vía biliar intrahepática, mientras que la colangiopancreatografía retrógrada endoscópica contribuye a la instrumentación de la vía biliar, a la extracción de los cálculos en ella y a la toma de biopsias ante la sospecha de lesiones neoplásicas de los conductos. Asimismo, ayudan a la dilatación de la estenosis de la vía biliar y a la instalación de endoprótesis para permeabilizar el colédoco, en casos de estrechez clínicamente manifiesta 10(Figura 3).

Tratamiento
La CBP y la CEP son los prototipos de las enfermedades crónicas colestásicas y, por tanto, se consideran los trastornos modelo para discutir el manejo médico de la colestasis 48. Las estrategias de tratamiento buscan limitar la acumulación de ácidos biliares y reducir la reserva de estos, y apuntan a la hepatoprotección a partir de inducir coleresis o la excreción biliar de dichos ácidos, lo cual limita el daño del colangiocito y modula la inflamación generada por los ácidos biliares 49.
Así pues, el manejo de las enfermedades colestásicas (principalmente la CBP y la CEP) se sustenta en el uso de ácido ursodesoxicólico, el cual estimula el flujo biliar y de bicarbonato en los hepatocitos y en los colangiocitos. Además, brinda un efecto antiapoptótico y antiinflamatorio 50 para identificar los criterios de respuesta, luego del inicio de dicho tratamiento en la CBP 51) (Tabla 1).
Tabla 1 Criterios de respuesta luego del tratamiento de la CBP.

ALP: fosfatasa alcalina (Alkaline Phosphatase); AST: aspartato aminotransferasa (Aspartate Aminotransferase). Modificada de la referencia 51.
Sin embargo, en la CEP no existe suficiente evidencia que demuestre un beneficio para la supervivencia de los pacientes, pese a una mejoría en el perfil hepático 23. Otros agentes que aún se encuentran en uso son los agonistas de los receptores farnesoide X (Farnesoid X Receptor, FXR) -como el ácido obeticólico-, el receptor retinoide X (Receptor X Retinoide, RXR), el receptor pregnano X (Pregnane X Receptor, PXR), el receptor de glucocorticoide (GR) y el receptor a del proliferador activado de peroxisomas (Perosyxomel Proliferator Activated Receptors, PPAR a).
En estos receptores actúan los fibratos como el fenofibrato y el bezafibrato, los cuales son modificadores transcripcionales de la formación de bilis 52 y previenen la acumulación de ácidos biliares, lo que reduce la importación y el aumento de la exportación de estas moléculas en los hepatocitos 49.
En caso de que se presenten pacientes sin respuesta al uso de medidas farmacológicas, se debe considerar el trasplante hepático, el cual alcanza supervivencias a 1 y 5 años del 83 y el 78 %, respectivamente. No obstante, existe la posibilidad de que haya recurrencia de la enfermedad, la cual puede alcanzar hasta un 8 % luego de 5 años del trasplante, y un 22 % a los 10 años de haber realizado el procedimiento 24.
Conclusiones
La colestasis es un síndrome que compromete la síntesis, la secreción y el flujo de la bilis y puede derivar en riesgo de cirrosis e hipertensión portal. Es importante su diagnóstico temprano y la identificación de manifestaciones sutiles como el prurito y los cambios en los niveles de ALP, para lo cual es necesario tener claridad del origen hepático y excluir otras fuentes, dada su presencia en otros tejidos.
Así pues, el tratamiento busca disminuir la inflamación generada por los ácidos biliares y modificar la reserva de estos, bien sea por su excreción o por el bloqueo de la circulación enterohepática. En consecuencia, es fundamental excluir las complicaciones causadas por esta alteración como la presencia de osteoporosis y la deficiencia de vitaminas, y tratarlas oportunamente.

