











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Copper is an essential trace element in living organisms and its consumption exceeds the baseline requirement1. ATP7B protein, which is found in hepatocytes, is responsible for the excretion of copper through bile and adds it to apoceruloplasmin for its distribution2,3. In Wilson’s disease, mutations in this protein leads to copper accumulation, which in turn causes multiple organ dysfunction syndrome. It has an autosomal recessive inheritance pattern and, currently, more than 600 pathogenic mutations have been described4,5. In Colombia, Wilson’s disease is considered an orphan disease6. Worldwide, a prevalence of 0.5 cases per 100 000 inhabitants has been estimated, and its incidence in most populations is 1 per 30 000 live births7,8.
Its clinical presentation is highly variable and it can occur at any age, although its occurrence is more common in individuals aged between 5 and 35 years, with both acute and chronic symptoms and multiple organ involvement, being hepatic, neurological and psychiatric involvement predominant9,10. There is not a single test that allows confirming its diagnosis; in fact, several tests, along with clinical findings, are required to diagnose it.
Although its occurrence has been reported worldwide, it has been described that its prevalence is higher in closed areas with low genetic variability and high rates of inbreeding. In Latin America there are reports of isolated cases and small case series in most countries of the region; however, the experience of Costa Rica stands out, as in said country there are areas in which the incidence of Wilson’s disease can reach up to 60 cases per 100,000 inhabitants11. Like in other Latin American countries, only isolated cases have been reported in Colombia, however information regarding the behavior of this disease in the country is insufficient12,13.
The aim of this study is to describe a retrospective case series, identifying the clinical characteristics, diagnostic tests, treatment plans and outcomes in patients with Wilson’s disease treated at our health center.
Materials and methods
Data collection
Information was obtained retrospectively from the database of the gastroenterology and hepatology group of the Universidad de Antioquia and the Hospital Pablo Tobón Uribe (HPTU) in Medellín, Colombia. The medical records with the diagnostic code E830 (disorder of copper metabolism, unspecified) of patients treated between January 2004 and September 2017 were searched without any restriction.
Diagnostic criteria
The medical records of 50 patients were collected and those who did not meet the Leipzig diagnostic criteria for Wilson’s disease were excluded from the analysis. Leipzig criteria include the presence of Kayser-Fleischer rings, serum ceruloplasmin level, neurological involvement, presence of non-immune hemolytic anemia, quantification or detection of copper in liver biopsy, urinary copper, and the detection of pathogenic mutations14. All patients included scored more than 4 points according to this diagnostic scale (Table 1).
Table 1 Scoring system developed at the 8th International Meeting on Wilson’s Disease, Leipzig, 2001
| Symptoms, signs and tests | Points | Symptoms, signs and tests | Points |
|---|---|---|---|
| Kayser-Fleischer rings: | Liver copper (in the absence of cholestasis): | ||
| - Present | 2 | - 5 x LSN (> 4 μmol/g) | 2 |
| - Absent | 0 | - 0,8-4 μmol/g | 1 |
| - Normal (< 0.8 μmol/g) | -1 | ||
| - Rhodamine-positive granules* | 1 | ||
| Neurological symptoms:** | Urinary copper (in the absence of acute hepatitis): | ||
| - Severe | 2 | - Normal | 0 |
| - Mild | 1 | - 1-2 x LSN | 1 |
| - Absent | 0 | - > 2 x LSN | 2 |
| - Normal, but > 5 x ULN after D-penicillamine | 2 | ||
| Serum ceruloplasmin: | Mutation analysis: | ||
| - Normal (> 0.2 g/L) | 0 | - Detected on both chromosomes | 4 |
| - 0,1-0,2 g/L | 1 | - Detected on 1 chromosome | 1 |
| - < 0,1 g/L | 2 | - No mutations detected | 0 |
| Coombs-negative hemolytic anemia: | |||
| - Present | 1 | ||
| - Absent | 0 | ||
| Total score | Result of the evaluation | ||
| 4 or more | Confirmed | ||
| 3 | Possible | ||
| 2 or less | Unlikely | ||
*If no quantitative liver copper available. **If there are no typical abnormalities at brain MRI. ULN: upper limit of normality.
Variables
Categorical variables were analyzed in all patients, including general demographic variables, presence of comorbidities, age at symptom onset, age at diagnosis, type of presentation, characteristics of laboratory tests, imaging studies, histopathological findings, follow-up time, treatment and outcomes. Diagnostic tests, imaging and genetic studies are described according to their availability. Likewise, the criteria for determining clinical and biochemical improvement, stability and survival were based on the data recorded by the treating hepatologist in the medical records.
Statistical analysis
Data collection and analysis were performed using Epidat 4.2 software. A descriptive analysis of all patients with Wilson’s disease was carried out.
Quantitative variables are expressed using means or medians with their respective measures of dispersion depending on the data distribution of each variable.
Results
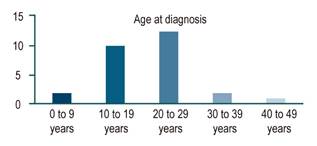
After reviewing the medical records, 27 patients met the diagnostic criteria for Wilson’s disease: 17 (62 %) were men and 10 (37 %), women. In addition, 24 (88 %) were from municipalities of the department of Antioquia. Average age of patients at diagnosis was 21.2 years (8 to 42 years). Young age was predominant in the sample, as 81% were between the ages of 10 and 30 years (Figure 1).
Clinical presentation of the disease was heterogeneous and symptoms overlapped in many patients (Figure 2). Liver involvement was the most frequent clinical presentation, followed by neurological and psychiatric involvement.
Liver involvement
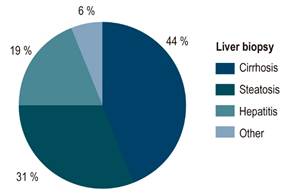
Liver involvement was reported in 23 (85 %) patients: cirrhosis (55 %), steatosis (14.81 %), acute liver failure (7.41 %) and asymptomatic biochemical alteration (7.41 %). A liver biopsy was performed in 16 patients, being cirrhosis (44 %), steatosis (31 %) and chronic hepatitis (19 %) the most frequent findings. Copper deposits in special stains were documented in only 7 patients (Figure 3). Acute liver failure was reported in two women aged 22 and 25 years; besides, this was the initial clinical manifestation of the disease in both cases, none of them experienced neuropsychiatric manifestations. Biochemical presentation was typical, with severe hyperbilirubinemia and low alkaline phosphatase; one patient presented with acute renal failure and required renal replacement therapy. Both patients underwent cadaveric donor liver transplantation and had a good outcome.
Neurological involvement
Neurological involvement was found in 11 patients (40 %) as follows: parkinsonism (14 %), ataxia (7.4 %), dystonia (11 %) and tremor (7 %).
Psychiatric involvement
Psychiatric symptoms were reported in 8 patients (29 %), being depression, anxiety, and schizophrenia the most frequent.
Other manifestations associated with Wilson’s disease
Four patients developed Coombs-negative hemolytic anemia: in two cases it was associated with acute liver failure, and in the remaining two it was associated with acute-on-chronic liver failure. Also, hypogonadism was reported in one patient (Table 2).
Table 2 Clinical presentation
| Clinical presentation | n (%) |
|---|---|
| Average age in years | 21 (8-42) |
| Sex | |
| - Male | 17 (62) |
| - Female | 10 (37) |
| Liver involvement | 23 (85) |
| - Cirrhosis | 15 |
| - Steatosis | 4 |
| - Acute liver failure | 2 |
| - Biochemical alteration | 2 |
| Neurological involvement | 11 (40) |
| - Parkinsonism | 4 |
| - Ataxia | 2 |
| - Dystonia | 3 |
| - Tremor | 2 |
| Psychiatric involvement | 8 (29) |
| Asymptomatic | 6 (22) |
Laboratory tests
Regarding laboratory tests, low ceruloplasmin levels and high urinary copper levels were reported in 23 (85 %) and 15 (55 %) patients, respectively. The 4 patients with acute presentation (2 with acute liver failure and 2 with acute-on-chronic liver failure) had positive results in the alkaline phosphatase/total bilirubin (in which positivity is defined as having a score < 4) and alanine aminotransferase (ALT)/aspartate aminotransferase (AST) (in which positivity is defined as having a score > 2.2) ratios. Kayser-Fleisher rings were found in 11 patients (40 %) during slit lamp exam. Finally, 12 MRIs and 2 CT scans of the central nervous system (CNS) were performed, and findings suggestive of Wilson’s disease were reported in 9 patients (86 %) (Table 3).
Table 3 Clinical presentation and laboratory tests
| Laboratory test | Clinical presentation | ||
|---|---|---|---|
| Liver (n: 24) | Neurological (n: 11) | Psychiatric (n: 8) | |
| Low ceruloplasmin < 0.2 g/L | 20 (83 %) | 9 (81 %) | 6 (75 %) |
| Urinary copper | 15 (62,5 %) | 6 (54,5 %) | 3 (37,5 %) |
| Kayser-Fleisher ring | 7 (29,1 %) | 8 (72,7 %) | 4 (50 %) |
| Copper on liver biopsy | 7 (29 %) | 0 (0 %) | 2 (25 %) |
Genetic testing was performed with confirmatory results in 3 patients. It was considered that the mutation detected in these patients is a homozygous variant (c.3694 A > C, missense mutation), probably pathogenic, that substitutes threonine for proline at position 1232 of the protein and affects various functional domains). Table 3 shows the distribution of positive diagnostic tests in the different clinical settings (liver, neurological and psychiatric involvement).
Treatment
Regarding treatment, 15 patients (55 %) were administered zinc and D-penicillamine (55 %), 3 (11 %), only zinc, and 3 (11 %), only D-penicillamine. Liver transplantation was required in 6 cases (22 %; 2 patients with liver failure without medical treatment and 4 with decompensated cirrhosis). There were 2 adverse drug reactions, one of them severe to D-penicillamine (febrile neutropenia), so trientine hydrochloride administration was started as a second-line drug in this patient.
During follow-up, 14 patients (51.8 %) showed clinical or biochemical improvement; 3 remained stable (11 %) and in 6 their condition worsened (18 %). There was no follow-up information of 4 patients. Of the total number of patients, 24 survived during follow-up (88 %) and 4 died.
Discussion
This case series reports new data on Wilson’s disease in Colombia. One of the most relevant findings is that 24 patients were from Antioquia, having aggregation in subregions of the department; this is due to the founder effect in these isolated subregions of Antioquia, where the poor genetic variability given by consanguinity leads to the expression of diseases with recessive Mendelian inheritance. This theory is supported by the fact that the same mutation was detected in three patients who underwent genetic testing, despite they did not have any family relationship.
The age peak of Wilson’s disease onset is between 10 and 30 years in both men and women, which shows that it mainly affects young people; in addition, its late detection is a socio-economic tragedy for the families of these patients, as their working life is reduced, which, in turn, perpetuates the socio-economic gap.
Wilson’s disease has been described as a heterogeneous disease, both clinically and genetically15. In our case series this was also the case; besides, overlapping between the different symptoms was highly frequent. Regarding liver involvement, liver cirrhosis was the most frequent manifestation, while biochemical alteration alone and acute liver failure were the less frequent. An important histopathological finding was that only steatosis was reported in 31% of the biopsies, a finding that can be identified in more common liver disorders, thus, Wilson’s disease must be also suspected in compatible clinical settings. These data are similar to what has been described in other case series, where liver involvement is predominant (58%-83%) and cirrhosis is the most frequent clinical presentation (31%)9. Compared to our study, in one of such case series, steatosis was more frequently found in renal biopsies (54%), while cirrhosis was only reported in 37%10. The late diagnosis of Wilson’s disease and the deferral of liver biopsy in patients with more advanced clinical manifestations and symptoms are a possibility in our region.
All patients with hemolytic anemia presented with acute liver failure or acute-on-chronic liver failure; this explains the occurrence of hemolysis in Wilson’s disease, caused by the excessive release of copper ions into the blood when hepatocytes are injured16,17. Also, the 4 patients with these conditions had scores typical of acute liver failure in Wilson’s disease in the AP/TB (< 4) and AST/ALT (> 2.2) ratios, which confirms their usefulness and their high sensitivity and specificity rates in this clinical setting18.
Endocrine involvement was observed in one of the patients: hypogonadism associated with decreased somatomedin C levels and elevated growth hormone levels. Hypogonadotropic hypogonadism has been described in patients with Wilson’s disease, but this finding is possibly associated with the progression of liver disease19.
Different diagnostic methods were used in all patients, which proves the importance of the Leipzig criteria14. As shown in Table 3, not all patients with the two most frequent clinical presentations (liver and neurological involvement) had low ceruloplasmin levels or elevated urinary copper levels. In general, the diagnostic process of this disease is complex and requires both extensive clinical experience and adequate interpretation of diagnostic aids.
Basal ganglia T2 hyperintensity was the most frequent finding in neuroimaging tests, while the classic finding of Wilson’s disease (panda sign of the midbrain) was only described in one patient. Interestingly, T1 hyperintensity was observed in four patients; this is a reflection of manganese accumulation associated with cirrhosis or liver failure20. In addition, cortical atrophy, leukoaraiosis and ventriculomegaly was a finding reported in both MRI and CAT scan, which suggests that there are clinical presentations with a more diffuse involvement, besides the possible cognitive involvement of these patients, which has been evidenced in different studies21.
Response to medical treatment was generally good. Of the patients who were treated with zinc plus D-penicillamine, 86 % experienced clinical improvement; 6 %, stabilization of their clinical condition, and in only 1, clinical condition worsened until cirrhosis was developed, thus requiring liver transplantation. In this regard, a multicenter cohort study carried out in Germany and Austria reported that patients with liver presentation of Wilson’s disease experienced clinical improvement with pharmacological treatment: 91% of those treated with D-penicillamine and 92% of those who were administered trientine22. A similar cohort study found that therapeutic failure was more frequent in patients treated with zinc (15 %), compared to those treated with chelating agents (1.2 %)23. In our study, only 6 patients showed a worsened condition and 4 died during follow-up; of these, one died due to immunosuppressive treatment discontinuation after undergoing liver transplantation and the other 3 due to infection-related complications during hospitalization. Overall, a survival rate of 85% was reported during follow-up, so it can be concluded that good outcomes can be achieved in patients with Wilson’s disease, as long as it is early detected and properly treated according to its clinical stage.
This is a retrospective observational study and, as such, it has the limitations inherent to this methodology. However, it is an important tool for describing the behavior of Wilson’s disease in our region and clearly identifying a more frequent geographic area of the disease associated with the founder effect and endogamy. This work serves as a starting point for future population-based studies, with the purpose of detecting a greater number of cases, achieving early confirmation of Wilson’s diagnosis or diagnosing it in preclinical phases to improve prognosis and, in addition, conducting genetic counseling campaigns in the region.

