










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev. colomb. Gastroenterol. vol.36 no.1 Bogotá Jan./Mar. 2021 Epub Oct 21, 2021
https://doi.org/10.22516/25007440.593
Artículos originales
Enfermedad de Wilson: experiencia de un centro de referencia en Colombia

1Clinical Hepatology and Gastroenterology Group of Universidad de Antioquia. Hospital Pablo Tobón Uribe. Medellín, Colombia.
2MD, surgeon. Clinical Hepatology and Gastroenterology Group. Universidad de Antioquia. Medellín, Colombia.
Introducción:
La enfermedad de Wilson es una enfermedad heterogénea causada por mutaciones en el gen ATP7B. La presentación clínica es variable, en fenotipos hepáticos y neuropsiquiátricos. El objetivo de este estudio es describir una cohorte retrospectiva de pacientes.
Materiales y métodos:
Estudio retrospectivo descriptivo de pacientes atendidos en el Hospital Pablo Tobón Uribe desde enero de 2004 a septiembre de 2017.
Resultados:
Se reportaron 27 pacientes, 17 hombres y 10 mujeres. El tiempo de seguimiento medio fue de 2,18 años, el 40% presentó síntomas neurológicos; el 29%, psiquiátricos; y el 85%, alteración hepática. En el laboratorio, el 85% presentó ceruloplasmina baja; 55%, cobre urinario alto; en casos con biopsia hepática, 7 tenían depósito de cobre en coloraciones especiales. En neuroimágenes, el 84% presentó hallazgos sugestivos de enfermedad de Wilson y en 3 casos se documentó una mutación genética patogénica. Durante el seguimiento, el 51% mejoró clínica o bioquímicamente, el 11% se mantuvo estable y el 18% se deterioró. El 88% de los casos sobrevivió al final del seguimiento.
Conclusiones:
Este estudio es la cohorte retrospectiva más grande de Colombia. Los resultados son base para nuevos estudios poblacionales buscando de manera activa la enfermedad para documentarla en su fase preclínica y, de este modo, impactar en el pronóstico.
Palabras clave: Degeneración hepatolenticular; ATPasas transportadoras de cobre; cirrosis hepática; insuficiencia hepática aguda; proteína ATP7B humana; enfermedades de los ganglios basales
Introduction:
Wilson’s disease is a heterogeneous disorder caused by mutations in the ATP7B gene. Its clinical presentation is variable in hepatic and neuropsychiatric phenotypes. The aim of this study is to describe a retrospective cohort of patients.
Materials and methods:
A descriptive retrospective study was carried out in patients treated at the Hospital Pablo Tobón Uribe from January 2004 to September 2017.
Results:
27 patients were reported, 17 men and 10 women. The mean follow-up time was 2.18 years. 40 % of the patients had neurological symptoms, 29 % psychiatric symptoms, and 85 % hepatic impairment. Lab tests showed that 85 % had low ceruloplasmin and 55 % had increased urinary copper. In cases that underwent liver biopsy, 7 had special copper colorations. Neuroimaging revealed that 84 % had findings suggestive of Wilson’s disease and a pathogenic genetic mutation was documented in 3 cases. During follow-up, 51 % improved clinically or biochemically, 11 % remained stable, and 18 % deteriorated. 88 % of cases survived at the end of follow-up.
Conclusions:
This study is the largest retrospective cohort carried out in Colombia. The results are the basis for new population-based studies actively seeking this disease to describe its preclinical development and thus impact prognosis.
Keywords: Hepatolenticular degeneration; Copper-transporter ATPases; Liver cirrhosis; Acute liver failure; Human ATP7B protein; Basal ganglia diseases
INTRODUCCIÓN
El cobre es un oligoelemento esencial en los organismos vivos y su consumo sobrepasa la necesidad basal del mismo 1. La proteína ATP7B se encuentra en el hepatocito, se encarga de la excreción del cobre a través de la bilis y lo añade a la apoceruloplasmina para su distribución 2,3. Esta proteína se encuentra mutada en la enfermedad de Wilson, que lleva a la acumulación de cobre que genera una enfermedad multiorgánica. Actualmente, se han descrito más de 600 mutaciones patogénicas y su patrón de herencia es autosómico recesivo 4,5. En Colombia se considera como enfermedad huérfana 6, se estima a nivel mundial una prevalencia de 0,5 casos por 100 000 habitantes y su incidencia en la mayoría de las poblaciones es de 1 por 30 000 nacidos vivos 7,8.
La presentación clínica es muy variable y puede aparecer a cualquier edad, aunque es más común entre los 5 y 35 años, con sintomatología tanto aguda como crónica y afección multiorgánica, con predominio hepático, neurológico y psiquiátrico 9,10. No hay una prueba única para confirmar el diagnóstico, por lo cual se debe realizar un conjunto de pruebas que, asociadas con los hallazgos clínicos, conforman el diagnóstico.
A pesar de que su distribución es global, se han descrito zonas de mayor prevalencia, las cuales corresponden a zonas cerradas con poca variabilidad genética y altos índices de endogamia. En América Latina hay reportes de casos aislados y pequeñas series en la mayoría de los países; sin embargo, se debe resaltar la experiencia de Costa Rica. En este país hay zonas cuya incidencia puede llegar a ser de 60 casos por cada 100 000 habitantes 11. En Colombia ha ocurrido lo mismo que en otros países latinoamericanos, donde se reportan casos aislados, pero no hay información suficiente del comportamiento local 12,13.
El objetivo de este estudio es describir una serie de casos retrospectiva, identificando las características clínicas, pruebas diagnósticas, plan de manejo y desenlaces en pacientes con enfermedad de Wilson que fueron atendidos en nuestro centro.
MATERIALES Y MÉTODOS
Obtención de datos
Se obtuvo información retrospectiva de la base de datos del grupo de gastrohepatología de la Universidad de Antioquia y el Hospital Pablo Tobón Uribe (HPTU) en Medellín, Colombia. Se realizó la búsqueda de las historias clínicas de los pacientes con el código diagnóstico: E830: alteraciones en el metabolismo del cobre, desde enero de 2004 hasta septiembre de 2017, sin ningún tipo de restricción.
Criterios diagnósticos
Se recopilaron las historias clínicas de 50 pacientes y se excluyeron del análisis los pacientes que no cumplieran los criterios de Leipzig para enfermedad de Wilson. Los criterios de Leipzig incluyen la presencia del anillo de Kayser-Fleischer, el nivel de ceruloplasmina en el suero, el compromiso neurológico, la presencia de anemia hemolítica no inmune, la cuantificación o detección de cobre en la biopsia de hígado, el cobre urinario y el hallazgo de mutaciones patogénicas 14. Todos los pacientes tenían más de 4 puntos en esta escala diagnóstica (Tabla 1).
Tabla 1 Sistema de puntuación desarrollado en la 8.a Reunión Internacional Sobre la Enfermedad de Wilson, Leipzig, 2001
| Síntomas, signos y pruebas | Puntos | Síntomas, signos y pruebas | Puntos |
|---|---|---|---|
| Anillos de Kayser-Fleischer: | Cobre hepático (en ausencia de colestasis): | ||
| - Presentes | 2 | - 5 x LSN (> 4 μmol/g) | 2 |
| - Ausentes | 0 | - 0,8-4 μmol/g | 1 |
| - Normal (< 0,8 μmol/g) | -1 | ||
| - Gránulos rodamina positivos | 1 | ||
| Síntomas neurológicos:** | Cobre urinario (en ausencia de hepatitis aguda): | ||
| - Graves | 2 | - Normal | 0 |
| - Moderados | 1 | - 1-2 x LSN | 1 |
| - Ausentes | 0 | - > 2 x LSN | 2 |
| - Normal, pero > 5 x LSN después de la D-penicilamina | 2 | ||
| Ceruloplasmina sérica: | Análisis de mutaciones | ||
| - Normal (> 0,2 g/L) | 0 | - Detectadas en ambos cromosomas | 4 |
| - 0,1-0,2 g/L | 1 | - Detectadas en 1 cromosoma | 1 |
| - < 0,1 g/L | 2 | - Sin mutaciones detectadas | 0 |
| Anemia hemolítica Coombs negativa: | |||
| - Presente | 1 | ||
| - Ausente | 0 | ||
| Puntuación total | Resultado de la evaluación | ||
| 4 o más | Confirmado | ||
| 3 | Posible | ||
| 2 o menos | Poco probable | ||
*Si no hay cobre hepático disponible cuantitativamente. **Si no hay anomalías típicas en imágenes de resonancia magnética del cerebro. LSN: límite superior de la normalidad
Variables
En todos los pacientes se analizaron variables categóricas, entre ellas, variables demográficas generales, comorbilidades, edad de inicio de los síntomas, edad de diagnóstico, forma de presentación, características de laboratorio, imagenología, hallazgos histopatológicos, tiempo de seguimiento, tratamiento y desenlaces. Las pruebas diagnósticas, imágenes y estudios genéticos se describen según la disponibilidad; igualmente, los criterios de mejoría clínica, bioquímica, estabilidad y supervivencia se basaron en los datos consignados en la historia clínica por el hepatólogo tratante.
Análisis estadístico
La recolección y análisis de los datos se realizó con el software Epidat 4.2. Se realizó un análisis descriptivo de todos los pacientes con enfermedad de Wilson.
Las variables cuantitativas se expresan como media o mediana con sus respectivas medidas de dispersión, según la distribución de la variable.
RESULTADOS
En el registro de historias clínicas, 27 pacientes cumplieron los criterios diagnósticos de la enfermedad de Wilson: 17 (62%) eran hombres y 10 (37%), mujeres. 24 (88%) son procedentes de municipios del departamento de Antioquia. La edad promedio de los pacientes en el momento del diagnóstico fue de 21,2 años (8 a 42 años). Predominan las edades jóvenes, el 81% de la totalidad de pacientes estuvo entre los 10 y 30 años (Figura 1).
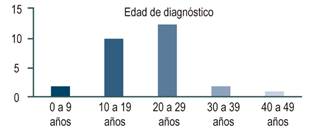
La presentación clínica fue heterogénea y muchos tenían solapamiento de la sintomatología (Figura 2). La presentación más común fue el compromiso hepático, seguido por el neurológico y, finalmente, el psiquiátrico.
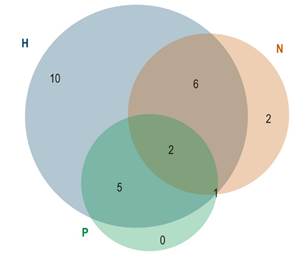
Figura 2 Síntomas iniciales de la enfermedad de Wilson. H: hepático; N: neurológico; P: psiquiátrico.
Compromiso hepático
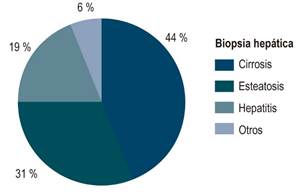
23 pacientes (85%) tuvieron compromiso hepático: cirrosis (55%), esteatosis (14,81%), insuficiencia hepática aguda (7,41%) y alteración bioquímica asintomática (7,41%). Se realizó una biopsia hepática en 16 pacientes y los hallazgos más frecuentes fueron cirrosis (44%), esteatosis (31%) y hepatitis crónica (19%). Solamente en 7 pacientes se documentó depósitos de cobre en las coloraciones especiales (Figura 3). Los casos de insuficiencia hepática aguda corresponden a dos mujeres de 22 y 25 años de edad que debutaron con esta presentación y no tuvieron manifestaciones neuropsiquiátricas. La presentación bioquímica fue típica, con hiperbilirrubinemia grave y fosfatasa alcalina baja; un caso se presentó con insuficiencia renal aguda y necesitó terapia de reemplazo renal. Ambas pacientes recibieron trasplante hepático con donante cadavérico y presentaron un buen desenlace.
Compromiso neurológico
Se encontró compromiso neurológico en 11 pacientes (40%), con las siguientes formas de presentación: parkinsonismo (14%), ataxia (7,4%), distonía (11%) y temblor (7%).
Compromiso psiquiátrico
8 pacientes (29%) tuvieron síntomas psiquiátricos. Los más frecuentes fueron depresión, ansiedad y esquizofrenia.
Otras manifestaciones asociadas
4 casos desarrollaron anemia hemolítica Coombs negativa, 2 fueron asociadas a insuficiencia hepática aguda, 2 a insuficiencia hepática aguda sobre crónica y un caso de hipogonadismo (Tabla 2).
Tabla 2 Presentación clínica
| Presentación clínica | n (%) |
|---|---|
| Promedio de edad en años | 21 (8-42) |
| Sexo | |
| - Masculino | 17 (62) |
| - Femenino | 10 (37) |
| Hepática | 23 (85) |
| - Cirrosis | 15 |
| - Esteatosis | 4 |
| - Insuficiencia hepática aguda | 2 |
| - Alteración bioquímica | 2 |
| Neurológica | 11 (40) |
| - Parkinsonismo | 4 |
| - Ataxia | 2 |
| - Distonía | 3 |
| - Temblor | 2 |
| Psiquiátrica | 8 (29) |
| Asintomático | 6 (22) |
Pruebas de laboratorio
En cuanto a pruebas de laboratorio, 23 pacientes (85%) tuvieron la ceruloplasmina baja y 15 (55%), cobre urinario alto. En los 4 pacientes con presentación aguda (2 con insuficiencia hepática aguda y 2 con insuficiencia hepática aguda sobre crónica) se realizaron los índices de fosfatasa alcalina/bilirrubina total (cuyos puntajes < 4 son positivos) y alanina-aminotransferasa (ALT)/aspartato-aminotransferasa (AST) (cuyos puntajes > 2,2 son positivos); todos presentaron estos índices positivos. En la evaluación oftalmológica con lámpara de hendidura se encontraron 11 pacientes con anillo de Kayser-Fleisher (40% de los pacientes). Se realizaron 12 resonancias magnéticas y 2 tomografías simples del sistema nervioso central (SNC), y hubo hallazgos sugestivos de enfermedad de Wilson en 9 pacientes (86%) (Tabla 3).
Tabla 3 Presentación clínica y laboratorios
| Laboratorio | Presentación clínica | ||
|---|---|---|---|
| Hepática (n: 24) | Neurológica (n: 11) | Psiquiátrica (n: 8) | |
| Ceruloplasmina baja < 0,2 g/L | 20 (83 %) | 9 (81 %) | 6 (75 %) |
| Cobre urinario | 15 (62,5 %) | 6 (54,5 %) | 3 (37,5 %) |
| Anillo de Kayser-Fleisher | 7 (29,1 %) | 8 (72,7 %) | 4 (50 %) |
| Cobre en biopsia hepática | 7 (29 %) | 0 (0 %) | 2 (25 %) |
Se realizó una prueba genética con resultado confirmatorio en 3 pacientes, la mutación encontrada se considera una variante probablemente patogénica, homocigota (c.3694 A > C, mutación de sentido erróneo que sustituye treonina por prolina en la posición 1232 de la proteína y afecta diversos dominios funcionales). En la Tabla 3 se puede observar la distribución de las pruebas diagnósticas positivas en los diferentes escenarios clínicos (compromiso hepático, neurológico y psiquiátrico).
Tratamiento
Con respecto al tratamiento, 15 pacientes recibieron zinc y D-penicilamina (55%), 3 recibieron solamente zinc (11%) y 3 únicamente D-penicilamina (11%). 6 pacientes requirieron trasplante hepático (22%; 2 pacientes con insuficiencias hepáticas sin tratamiento médico y 4 cirróticos descompensados). Se registraron 2 reacciones adversas, 1 de ellas grave a la D-penicilamina (neutropenia febril), por lo cual en este caso se inició como segunda línea el clorhidrato de trientina.
Durante el seguimiento, 14 pacientes (51,8%) presentaron mejoría clínica o bioquímica; 3 se mantuvieron estables (11%) y 6 se deterioraron (18%). En 4 pacientes no se encontró información sobre el seguimiento. Del total de pacientes, 24 sobrevivieron durante el seguimiento (88%) y 4 fallecieron.
DISCUSIÓN
En esta serie de casos se reportan nuevos datos de la enfermedad de Wilson en Colombia. Uno de los hallazgos más importantes es que 24 pacientes eran procedentes de Antioquia, teniendo agregación en subregiones del departamento, esto se debe al efecto fundador en estas subregiones aisladas de Antioquia, donde la pobre variabilidad genética dada por la consanguinidad lleva a la expresión de enfermedades con herencia mendeliana recesiva. Esta teoría se soporta en que 3 pacientes a quienes se les realizó estudio genético presentaron la misma mutación, sin estar familiarmente relacionados.
El pico de edad en ambos sexos es entre los 10 a 30 años, lo que demuestra que esta es una enfermedad de personas jóvenes; y la identificación tardía es una tragedia socioeconómica para las familias, puesto que se disminuye el tiempo de vida laboral productiva, lo que perpetúa la brecha socioeconómica.
Se ha descrito la enfermedad de Wilson como una enfermedad heterogénea, tanto clínica como genéticamente 15. Se puede evidenciar que no es diferente en nuestra serie y se encontró gran solapamiento entre las diferentes sintomatologías. Con respecto al compromiso hepático, se evidenció como manifestación más común la cirrosis hepática, y las presentaciones menos frecuentes fueron la alteración bioquímica únicamente y la insuficiencia hepática aguda. Un hallazgo importante en la histopatología fue que el 31% de las biopsias presentó únicamente esteatosis, hallazgo que se puede encontrar en patologías hepáticas más comunes, lo que obliga en el marco clínico compatible siempre pensar también en enfermedad de Wilson. Estos datos son similares a otras series, en las que hubo predominancia hepática (58%-83%) y la cirrosis fue la presentación más frecuente (31%) 9. En la biopsia hepática de una de las series, se encontró mayor cantidad de esteatosis (54%) y cirrosis únicamente en el 37% 10. Es posible que en nuestra región se esté diagnosticando de manera tardía la enfermedad o que se esté difiriendo la biopsia hepática en pacientes con clínica más avanzada.
Todos los pacientes con anemia hemolítica se presentaron con insuficiencia hepática aguda o insuficiencia hepática aguda sobre crónica; esto corrobora la explicación de la hemólisis en la enfermedad de Wilson, causada por la liberación excesiva de iones de cobre en la sangre al lesionarse los hepatocitos 16,17. También se evidenció cómo los 4 pacientes presentaron los índices clásicos de Wilson en insuficiencia hepática aguda (FA/BT < 4 y AST/ALT > 2,2), lo que corrobora su utilidad y sus altos índices de sensibilidad y especificidad en este escenario 18.
Uno de los pacientes presentó compromiso endocrinológico: tuvo hipogonadismo asociado con la disminución de somatomedina C con hormona del crecimiento elevada. Se ha encontrado hipogonadismo hipogonadotrópico en pacientes con enfermedad de Wilson, pero este hallazgo posiblemente esté asociado con lo avanzado de la hepatopatía 19.
En todos los pacientes se utilizaron diferentes métodos diagnósticos, lo que demostró la importancia de los criterios de Leipzig 14. Como se puede observar en la Tabla 3, en ninguna de las dos presentaciones más comunes (hepática y neurológica) se encontró en el 100% de las veces ceruloplasmina baja o cobre urinario elevado. El proceso diagnóstico en general es complejo y exige una amplia experiencia clínica y una adecuada interpretación de las ayudas diagnósticas.
Dentro de las pruebas de neuroimagen, se encontró como hallazgo más prevalente la hiperintensidad en T2 en los ganglios basales y el hallazgo clásico de enfermedad de Wilson (el oso panda en el mesencéfalo y tegmento) únicamente en un paciente. De manera interesante, se encontraron 4 pacientes con hiperintensidad en T1; esto es reflejo de la acumulación de manganeso asociado con cirrosis o insuficiencia hepática 20. Además, un hallazgo encontrado tanto en resonancia magnética como en tomografía axial computarizada (TAC) fue la atrofia cortical generalizada, leucoaraiosis y ventriculomegalia, por lo cual se puede plantear que hay presentaciones con un compromiso más difuso, además del posible compromiso cognitivo de los pacientes con esta enfermedad, el cual ha sido evidenciado en diferentes estudios 21.
La respuesta al tratamiento médico en general es buena. De los pacientes que recibieron tratamiento combinado de zinc y D-penicilamina, el 86% presentó mejoría; el 6%, estabilización de su estado clínico y solo 1 caso de ellos progresó su estadio de cirrosis y requirió trasplante hepático. En una cohorte multicéntrica realizada en Alemania y Austria se encontró mejoría con tratamiento médico, en pacientes con presentación hepática en un 91% tratados con D-penicilamina y en un 92%, con trientina 22. En una cohorte similar se encontró que era más común encontrar falla terapéutica en pacientes manejados con zinc (15%), comparados con pacientes manejados con quelantes (1,2%) 23. En el seguimiento se puede ver cómo solo 6 pacientes se deterioraron y 4 murieron; de estos, 1 murió por la descontinuación del tratamiento inmunosupresor postrasplante hepático y los otros 3 por complicaciones infecciosas durante la hospitalización. En general, se reportó una supervivencia del 85% durante el seguimiento, por lo que se puede concluir que la enfermedad de Wilson puede tener buenos desenlaces, mientras se identifique a tiempo y se trate de acuerdo con el estadio clínico.
Al ser un estudio observacional retrospectivo, tiene las limitaciones propias de la metodología. Sin embargo, es una herramienta importante para describir el comportamiento de la enfermedad de Wilson en nuestra región e identificar de manera clara un área geográfica con mayor frecuencia de la enfermedad asociada con el efecto fundador y la endogamia. Esto sirve como punto de partida para la realización de estudios poblacionales en el campo, con el ánimo de detectar un mayor número de casos, realizar la confirmación diagnóstica temprana o en fases preclínicas para mejorar el pronóstico y, adicionalmente, para realizar campañas de asesoría genética en la región.
REFERENCIAS
1. Pohanka M.. Copper and copper nanoparticles toxicity and their impact on basic functions in the body. Bratisl Lek Listy. 2019;120(6):397-409. https://doi.org/10.4149/BLL_2019_065 [ Links ]
2. Broussolle E, Trocello JM, Woimant F, Lachaux A, Quinn N. Samuel Alexander Kinnier Wilson. Wilson’s disease, Queen Square and neurology. Rev Neurol (Paris). 2013;169(12):927-35. https://doi.org/10.1016/j.neurol.2013.04.006 [ Links ]
3. Harada M, Kawaguchi T, Kumemura H, Terada K, Ninomiya H, Taniguchi E, Hanada S, Baba S, Maeyama M, Koga H, Ueno T, Furuta K, Suganuma T, Sugiyama T, Sata M. The Wilson disease protein ATP7B resides in the late endosomes with Rab7 and the Niemann-Pick C1 protein. Am J Pathol. 2005;166(2):499-510. https://doi.org/10.1016/S0002-9440(10)62272-9 [ Links ]
4. Gomes A, Dedoussis GV. Geographic distribution of ATP7B mutations in Wilson disease. Ann Hum Biol. 2016;43(1):1-8. https://doi.org/10.3109/03014460.2015.1051492 [ Links ]
5. Trocello JM, Broussolle E, Girardot-Tinant N, Pelosse M, Lachaux A, Lloyd C, Woimant F. Wilson’s disease, 100 years later…. Rev Neurol (Paris). 2013;169(12):936-43. https://doi.org/10.1016/j.neurol.2013.05.002 [ Links ]
6. Resolución 2048 de 2015, por la cual se actualiza el listado de enfermedades huérfanas y se define el número con el cual se identifica cada una de ellas en el sistema de información de pacientes con enfermedades huérfanas. Ministerio de salud y Protección Social, Republica de Colombia (9 de junio de 2015). [ Links ]
7. Kim GH, Yang JY, Park JY, Lee JJ, Kim JH, Yoo HW. Estimation of Wilson’s disease incidence and carrier frequency in the Korean population by screening ATP7B major mutations in newborn filter papers using the SYBR green intercalator method based on the amplification refractory mutation system. Genet Test. 2008;12(3):395-9. https://doi.org/10.1089/gte.2008.0016 [ Links ]
8. Kumar S, Thapa B, Kaur G, Prasad R. Analysis of most common mutations R778G, R778L, R778W, I1102T and H1069Q in Indian Wilson disease patients: correlation between genotype/phenotype/copper ATPase activity. Mol Cell Biochem. 2007;294(1-2):1-10. https://doi.org/10.1007/s11010-005-9028-z [ Links ]
9. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut. 2007;56(1):115-20. https://doi.org/10.1136/gut.2005.087262 [ Links ]
10. Cheung KS, Seto WK, Fung J, Mak LY, Lai CL, Yuen MF. Epidemiology and natural history of Wilson’s disease in the Chinese: A territory-based study in Hong Kong between 2000 and 2016. World J Gastroenterol. 2017;23(43):7716-7726. https://doi.org/10.3748/wjg.v23.i43.7716 [ Links ]
11. Hevia-Urrutia, F., Alvarado-Echeverría, I., Sanabria-Castro, A. Sánchez-Molina M, Meza-Sierra L, Parajeles-Vindas A, Méndez-Blanca O, Sánchez-Siles Á, Saborío-Rocafort M, Barguil-Gallardo M, Chavarría-Quirós I, Monge-Bonilla C. National alliance for Wilson’s disease: health policy in Costa Rica. Hepatology Medicine Policy 2017;2:5. https://doi.org/10.1186/s41124-016-0012-x [ Links ]
12. Pulido JN, Medina LA, De Castro A, Tamayo JG, Medina S, Restrepo JC. Enfermedad de Wilson. Acta Med Colomb. 2002;27:186-188. [ Links ]
13. Castaño O, Gómez DM, Ocampo JM, Casanova ME. Degeneración hepatolenticular: a propósito de tres casos. Iatreia. 2017;30(4):436-442. http://dx.doi.org/10.17533/udea.iatreia.v30n4a07 [ Links ]
14. Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23(3):139-42. https://doi.org/10.1034/j.1600-0676.2003.00824.x [ Links ]
15. Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9(2):210-7. https://doi.org/10.1038/ng0295-210 [ Links ]
16. Kiss JE, Berman D, Van Thiel D. Effective removal of copper by plasma exchange in fulminant Wilson’s disease. Transfusion. 1998;38(4):327-31. https://doi.org/10.1046/j.1537-2995.1998.38498257369.x [ Links ]
17. Hartleb M, Zahorska-Markiewicz B, Ciesielski A. Wilson’s disease presenting in sisters as fulminant hepatitis with hemolytic episodes. Am J Gastroenterol. 1987;82(6):549-51. [ Links ]
18. Korman JD, Volenberg I, Balko J, Webster J, Schiodt FV, Squires RH Jr, Fontana RJ, Lee WM, Schilsky ML; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology. 2008;48(4):1167-74. https://doi.org/10.1002/hep.22446 [ Links ]
19. Dzieżyc K, Litwin T, Członkowska A. Other organ involvement and clinical aspects of Wilson disease. Handb Clin Neurol. 2017;142:157-169. https://doi.org/10.1016/B978-0-444-63625-6.00013-6 [ Links ]
20. Yu XE, Gao S, Yang RM, Han YZ. MR Imaging of the Brain in Neurologic Wilson Disease. AJNR Am J Neuroradiol. 2019;40(1):178-183. https://doi.org/10.3174/ajnr.A5936 [ Links ]
21. Wenisch E, De Tassigny A, Trocello JM, Beretti J, Girardot-Tinant N, Woimant F. Cognitive profile in Wilson’s disease: a case series of 31 patients. Rev Neurol (Paris). 2013;169(12):944-9. https://doi.org/10.1016/j.neurol.2013.06.002 [ Links ]
22. Weiss KH, Thurik F, Gotthardt DN, Schäfer M, Teufel U, Wiegand F, Merle U, Ferenci-Foerster D, Maieron A, Stauber R, Zoller H, Schmidt HH, Reuner U, Hefter H, Trocello JM, Houwen RH, Ferenci P, Stremmel W; EUROWILSON Consortium. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11(8):1028-35.e1-2. https://doi.org/10.1016/j.cgh.2013.03.012 [ Links ]
23. Weiss KH, Gotthardt DN, Klemm D, Merle U, Ferenci-Foerster D, Schaefer M, Ferenci P, Stremmel W. Zinc monotherapy is not as effective as chelating agents in treatment of Wilson disease. Gastroenterology. 2011;140(4):1189-1198.e1. https://doi.org/10.1053/j.gastro.2010.12.034 [ Links ]
Citación: Muñoz-Maya O, Vélez-Hernández JE, Santos O, Marín JI, Restrepo-Gutiérrez JC. Enfermedad de Wilson: experiencia de un centro de referencia en Colombia. Rev Colomb Gastroenterol. 2021;36(1):51-57. https://doi.org/10.22516/25007440.593
Recibido: 12 de Junio de 2020; Aprobado: 23 de Noviembre de 2020
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
