text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
The first observations of patients with fatty liver date back to the 19th century1; nonalcoholic fatty liver disease (NAFLD), or fatty liver, as it is often called, was first described by Zelman in 1952 upon noting liver disease in very obese patients2. In 1980, Ludwig described the term nonalcoholic steatohepatitis (NASH) when observing a disease with histological changes similar to those in patients with alcoholic hepatitis but with negligible or absent alcohol use3, a concept that continues to be valid. Fatty liver is the leading cause of consultation in hepatology services. However, it is also frequent in consultations with clinical specialists and primary care physicians due, on the one hand, to changes in habits in modern life, hypercaloric diets, and a sedentary lifestyle, and on the other, greater access to the healthcare system. The increasing use of diagnostic aids leads to occasional findings of “fatty liver” on imaging (usually ultrasound) or elevated transaminases in asymptomatic patients4, which is why all doctors must be prepared to treat it. Therefore, these two topics are reviewed.
Definition and diagnosis
NAFLD, or fatty liver, is defined by the presence of fat or steatosis in hepatocytes and spans from the initial stage, simple steatosis without inflammation and fibrosis, to steatohepatitis (NASH) with inflammation and fibrosis, to cirrhosis, the most advanced stage5. MAFLD (Metabolic-Associated Fatty Liver Disease) has recently been proposed to encompass the metabolic alterations associated with fatty liver6. A consensus must be awaited regarding this new definition that partly emerges because the fatty liver is a highly heterogeneous disease with multiple subgroups.
Fatty liver is diagnosed by hepatic steatosis on imaging (usually ultrasound) or liver biopsy in 5% or more of the tissue examined, with or without inflammation or fibrosis. Secondary causes of hepatic steatosis should be ruled out, such as:
Alcohol use greater than 20 g/day for men and greater than 10 g/day for women;
Intake of hepatotoxic drugs in the last six months before the study;
Hepatitis B and C viruses, hemochromatosis, autoimmunity, and other causes of chronic liver disease5.
Epidemiology
Younossi showed a global prevalence close to 25% in the general population, with significant variations depending on the region of the world evaluated; the Middle East and South America showed the highest prevalence, 32%, and 31%, respectively, followed by North America (24%), Europe (23%), and Africa (13%)7. In the United States, other studies have reported NAFLD prevalences of 10% to 46%8,9. In Latin America, in a Mexican study with 2,503 individuals, NAFLD was detected in 14.3%, associated with overweight, obesity, and dyslipidemia10. In Chile, another study with 832 patients showed fatty liver in 23.4%11. In Colombia, we do not have exact prevalence data. Still, the 2015 National Nutritional Status Survey (ENSIN, for its acronym in Spanish) reported overweight in 19.1% of those surveyed between the ages of 13 and 17 and 38.4% of adults12.
Of note is that the prevalence of NAFLD has been increasing over time, as demonstrated by comparing three periods of the National Health and Nutrition Examination Survey (NHANES). Between 1988 and 1994, the prevalence of NAFLD (defined as elevated levels of serum aminotransferases with no explanation, which could result in underdiagnosis) was 5.5%; between 1999 and 2004, it was 9.8%, and between 2005 and 2008, it was 11%, representing 47%, 63%, and 75% of chronic liver diseases during those periods, respectively13.
Fatty liver is usually diagnosed between 40 and 50 years of age14, with variation regarding the predominant sex: in some studies, it is women3,15-17, and in others, men18-20.
There are ethnic differences in the prevalence of NAFLD18,21. A study of liver triglyceride content in 2,287 subjects of a multiethnic population sample from the United States found a higher prevalence of hepatic steatosis in Hispanic Americans (45%) compared with white (33%) or black (24%) people18. A higher prevalence of obesity explains the higher prevalence in Hispanics.
Fatty liver, especially NASH, is associated with metabolic syndrome (MS)5,20-23. Obesity is the leading risk factor since body mass index (BMI) and waist circumference positively correlate with fatty liver and disease progression22. Besides, 80% of patients are obese24,25), and 72% have dyslipidemia25-27. Type 2 diabetes (DM2) is found in 45% to 65% of patients with NAFLD and is directly and strongly associated with the severity and progression of fatty liver5,25-28. The combination of obesity, systemic hypertension, dyslipidemia, and insulin resistance or diabetes independently increases the risk of severe fibrosis and cardiovascular disease25,28-30.
Fatty liver is the most frequent liver disease in obese children and adolescents and is associated with chronic systemic disorders such as hypertension, dyslipidemia, and increased risk of DM2 and cardiovascular diseases. In the pediatric population, the prevalence of fatty liver is estimated to be between 7.6% and 34%31-33.
Fatty liver and obesity epidemics are global public health problems5,23,27, and fatty liver is the fastest-growing indication for liver transplantation due to decompensated cirrhosis or HCC34,35.
Pathophysiology
Fatty liver is a complex disorder and very heterogeneous pathophysiology resulting from the interaction of multiple genetic, epigenetic, environmental, and cultural factors. All this together produces hepatic fat accumulation, insulin resistance, hormonal alterations, and alterations in the intestinal microbiota, causing hepatocellular damage through the formation of oxygen-free radicals and activation of hepatic fibrogenesis.
Genetics
The first genome-wide association study in fatty liver (GWAS) showed the importance of heredity in the accumulation of liver fat and highlighted the susceptibility to the disease according to the individuals’ genetic status36. Subsequently, studies on twins confirmed the hereditary component of steatosis and fibrosis37. Other association studies reported genetic variants related to the risk of fatty liver, responsible for encoding proteins that regulate the metabolism of hepatic lipids that lead to the accumulation of liver fat. These variants are associated with the development and progression of fibrosis38,39, whose phenotypic expression is triggered by dietary factors and adiposity39. The most important genes and their variants or polymorphisms (SNPs) are presented below.
PNPLA3 gene
It encodes a phospholipase, adiponutrin, which regulates the metabolism of triglycerides and retinoids33,39. A single nucleotide polymorphism (SNP) of the gene, rs738409 (C > G), results in a sense variation (I148M) that inhibits the enzyme through a repressor that competitively binds to the adipocyte triglyceride lipase (ATGL) coactivator, causing a more significant accumulation of lipids (up to 75% more)36,40,41. Individuals with a G nucleotide variant have a 3.2 times higher risk of developing liver fibrosis, and NASH is more prevalent in GG individuals than in CC individuals (odds ratio [OR]: 3.49)41. The variant has been associated with steatohepatitis, fibrosis, and cancer. It has also been found to promote liver damage in alcoholic fatty liver, chronic hepatitis C, and infections that promote hepatic steatosis39.
The frequency of the PNPLA3-I148M variant was correlated with the ethnic origin and the prevalence of fatty liver in the population, being common in the mixed population (frequency of 26%), more prevalent in the Hispanic population (49%), and less prevalent in the African one (12-17%)36. In addition, the penetrance of this variant in the European population is comparable to the effects of the monogenic liver disease mutation, with homozygous GG having up to a 12-fold higher probability of developing HCC in patients with NAFLD42,43.
TM6SF2 gene or transmembrane 6 superfamily member 2
The gene regulates the lipid content of hepatocytes by encoding an endoplasmic reticulum (ER) transmembrane protein associated with the clearance of very low-density lipoproteins (VLDL)44. Its polymorphism (SNP), rs58542926 (G > A), resulting in the E167K variant, was associated with high levels of liver triglycerides and an increased risk of advanced fibrosis45. On the contrary, this variant decreases the secretion of VLDL by hepatocytes and reduces the risk of cardiovascular disease46.
MBOAT7 gene, membrane-bound O-acyltransferase domain-containing 7 (MBOAT7)
The MBOAT7 gene expression produces the enzyme lysophosphatidylinositol (LPI) acyltransferase, an endomembrane protein that catalyzes the production of phosphatidylinositol (PI), a component of cell membranes. Its SNP rs641738 has also been associated with increased fibrosis in fatty liver47,48.
GCKR gene, glucokinase regulatory gene (GCKR)
It is expressed in the liver and encodes a protein that acts as an allosteric inhibitor of GCK, the enzyme responsible for blood glucose homeostasis. Its SNP rs780094 genetic variant is associated with a higher fasting serum triacylglycerol level in fatty liver49.
HSD17B13 gene, 17β-Hydroxysteroid dehydrogenase (HSD17B13)
Its SNP rs72613567 is protective and reduces the risk of fatty liver; it was associated with reduced alanine aminotransferase (ALT)50,51.
Genetic basis of insulin resistance
It has been associated with polymorphisms in the apolipoprotein C3 gene, interleukin 6 (IL-6), and adiponutrin52-55, with altered transcriptional activity of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1A)52. Additionally, other cytokines and adipokines involved in insulin receptor signaling seem to be altered in the omental adipose tissue of NASH patients55.
Other genetic elements: non-coding RNA (ncRNA)
These RNAs do not code for functional proteins and are grouped according to their size, small (micro-RNA) and large, including long and circular RNAs. They regulate multiple biological pathways, lipid uptake, de novo lipogenesis, lipid oxidation, export of hepatic lipids, apoptosis, cell proliferation, or fibrosis56. At least a dozen micro-RNAs have been strongly associated with the development of fatty liver56,57. As cell death increases with the progression of steatohepatitis, they are released directly or packaged in exosomes into the circulation, making them potential biomarkers for fatty liver status58.
Excess lipid accumulation in the liver
The hepatic imbalance in the input and output of fats due to lifestyle changes caused by high-calorie consumption, sedentary lifestyle, metabolic syndrome, and hormonal and genetic alterations results in hepatic steatosis with excess triglycerides, free fatty acids (FFA), ceramides, and free cholesterol. It may occur due to excessive import of FFA from adipose tissue, decreased hepatic export of FFA (secondary to reduced VLDL synthesis or secretion), or impaired FFA β-oxidation. The primary sources of triglycerides are fatty acids stored in adipose tissue and newly processed fatty acids in the liver through de novo lipogenesis59,60. Additionally, de novo lipogenesis (DNL) has been reported to be upregulated in fatty liver patients. It is three-fold higher than in control patients, and this pathway may be pathogenetically more relevant in older patients and men61,62.
Increased visceral adipose tissue and intrahepatic fat correlate with increased gluconeogenesis, elevated FFA levels, and insulin resistance. Visceral fat has also been associated with liver inflammation and fibrosis in NASH patients independent of insulin resistance, an effect possibly mediated by IL-6 (proinflammatory cytokine)63,64.
Insulin resistance
It is one of the primary mechanisms in the pathophysiology of fatty liver; it occurs in most obese and diabetic patients, although it can also be found in lean non-diabetic patients65-67. Insulin resistance triggers multiple events in lipid metabolism: increased peripheral lipolysis and triglyceride synthesis, increased FFA uptake by the liver, and, thus, accumulation of triglycerides in hepatocytes, resulting in a preferential shift from carbohydrate to FFA β-oxidation65,68,69. The molecular pathways that lead to insulin resistance are complex and not fully elucidated, but several molecules appear to interfere with the actions of insulin at the cellular level; for example, lipophilic bile acids have been shown to promote insulin sensitivity and decrease gluconeogenesis and hepatic triglyceridemia by binding to the farnesoid X receptor70.
Gut microbiota
The human gut microbiome comprises 10 to 100 trillion microorganisms, primarily bacteria. It has ten times more intestinal microorganisms than human eukaryotic cells71 and is susceptible to environmental and pathophysiological alterations, which is why it plays a fundamental role in the pathophysiology of fatty liver. The microbiota has been associated with direct and indirect injury to the liver cell through various mechanisms that produce lipotoxicity, oxidative damage, and secondary fibrosis72-79:
Changes from the normal microbiota: bacterial overgrowth in the small intestine or changes in gut microbiota composition (for example, macronutrient composition in diets high in fructose and saturated fat).
Increased intestinal permeability possibly associated with small intestinal bacterial overgrowth, changes in microbiota composition, and bacterial translocation.
Increased production of endotoxins.
Generation of toxic products, such as endogenous alcohol and acetaldehyde by bacteria and yeast at the colon level, and other metabolites of the gut microbiota such as N,N,N-trimethyl-5-aminovaleric acid (TMAVA). The latter reduces the synthesis of carnitine and oxidation of hepatic fatty acids, leading to hepatic steatosis.
Deconjugation of bile salts and inactivation of hepatic lipotropes, such as choline.
In intestinal dysbiosis, defined as the imbalance between the resident microbial communities and the host, intestinal permeability increases may result in a pathological translocation of microbial products to the liver through the portal vein73-75. In this process, pathogen-associated molecular patterns (PAMPs) are produced, which are recognized by selective receptors in the liver, mainly Toll-like receptors, and provide a chronic innate immune response that releases damage-associated molecular patterns (DAMPs)80. Additionally, microbiota-derived metabolites, such as modified bile acids, choline, and ethanol, alter hepatic metabolism and trigger an inflammatory response72,75,77-80.
Peptides and hormones
Leptin
It is a peptide produced in adipose tissue; its absence is associated with massive obesity in mice (ob/ob) and humans. Leptin induces dephosphorylation of insulin receptor substrate 1, making hepatocytes more resistant to insulin81. It appears that leptin resistance in the central nervous system, rather than in the liver, may be necessary for the pathogenesis of NASH, which was inferred by observing that leptin infusion into the central nervous system of mice with fatty liver corrected insulin resistance and fatty liver, while peripheral administration did not82.
Adiponectin
It is another hormone secreted only in adipose tissue with beneficial effects on lipid metabolism. It improves β-oxidation of fatty acids in muscle and clearance of plasma lipids, inhibits tumor necrosis factor alpha (TNF-α) production in the liver, and produces direct anti-inflammatory effects83. Adiponectin appears to have some relationship with the modulation of insulin sensitivity, and low circulating hormone levels have been correlated with the severity of histopathological findings in NASH84.
Resistin
It is a protein derived from adipose tissue associated with developing insulin resistance. In research with mice, resistin overexpression led to altered FFA levels, glucose intolerance, and hyperinsulinemia85.
Incretins
These are gut-derived hormones, such as glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1), that potentiate insulin secretion after food intake and play an essential role in glucose homeostasis86.
Other proposed associated factors
Environmental factors
Modifiable risk factors, such as shift work and travel that disrupt normal eating and sleep-wake cycles, promote adiposity, metabolic syndrome, and nonalcoholic fatty liver disease87. Prolonged disruption of normal circadian rhythms in mice with fatty liver induces the development of nonalcoholic steatohepatitis by dysregulating crosstalk between two nuclear hormone receptors: the farnesoid X receptor (FXR) and the constitutive androstane receptor (CAR), resulting in FXR suppression, hepatic accumulation of bile acids, bile acid-induced CAR overactivation, and eventual CAR-dependent liver injury, fibrosis, and neoplasia88-90. Environmental toxins91 and obstructive sleep apnea, which cause inflammation, have also been implicated as pathogenic factors92,93.
Hepatocellular damage
It is ultimately produced by the formation of free radicals due to the following:
Induction of microsomal lipoxygenases of cytochrome p-450 from FFA;
The switch to β-oxidation of FFA plus pre-existing defects in mitochondrial oxidative phosphorylation (significant mitochondrial structural abnormalities), which have been demonstrated by electron microscopy of hepatocytes from NASH patients, not observed in patients with simple hepatic steatosis22,64-70.
These two pathways together lead to hepatocellular damage and fibrosis through the activation of multiple processes such as nuclear factor kappa B (NF-κB), increased production of cytokines, activation of TNF-α, the complement system, plasma myeloperoxidase, and natural killer cells65,94-96.
Oxygen-free radical formation and lipid peroxidation can deplete antioxidant enzymes such as glutathione, vitamin E, beta-carotene, and vitamin C, rendering the hepatocyte more susceptible to oxidative injury. In this process, serum levels of xanthine oxidase produced by reactive oxygen species are increased compared to control patients, while the levels of multiple antioxidant enzymes are decreased97,98.
A correlation has been described between the severity of the disease and the increased expression of oxidative scavenging receptors99. Serotonin has been implicated as a source of reactive oxygen species in NASH; increased serotonin catabolism resulted in increased levels of reactive oxidative species and necroinflammation in an animal model100.
Iron contributes to hepatocellular damage by forming oxygen-free radical species in its process of reducing Fe3+ to Fe2+(101, associated with the development of NASH; an increase in iron has also been observed in patients with insulin resistance independently102. A higher liver iron concentration correlates with the severity of fibrosis in NASH103.
The innate immune system plays a role in the progression of fatty liver104,105. An adequate physiological immune response is essential for resolving liver damage and normal liver regeneration. In contrast, a persistent and exaggerated response of the innate immune system may lead to chronic inflammation of the liver and its consequences. PAMPs and DAMPs activate and maintain a pattern-recognition receptor-mediated innate immune response characteristic of fatty liver and a complex intercellular crosstalk between different innate immune cells, natural killer cells, T lymphocytes, macrophages, neutrophils, hepatocytes, and hepatic stellate cells (HSCs). The result defines the progression to steatohepatitis and fibrosis105.
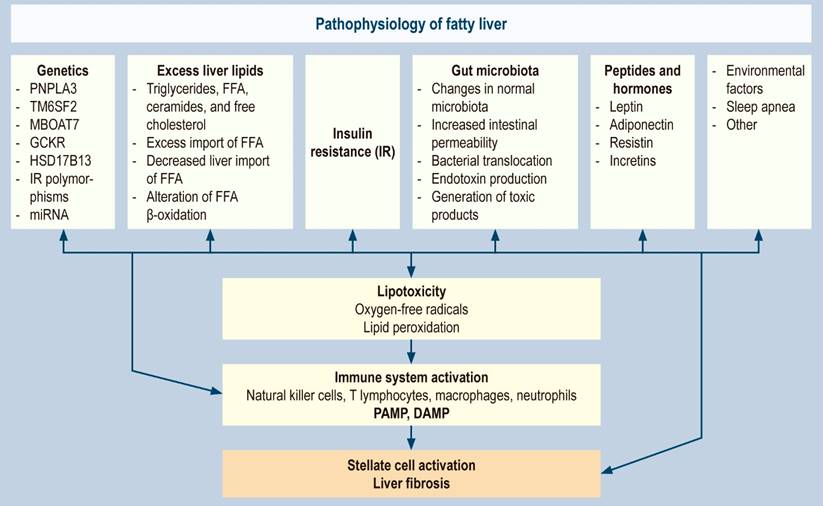
Apart from the death of hepatocytes by direct lipotoxicity and the release of DAMP, pyroptosis has been identified as a new form of programmed cell death in fatty liver, characterized by the activation and assembly of multiprotein complexes, called inflammasomes106,107. The most studied is the nucleotide-binding domain, a leucine-rich repeats (NLR) family pyrin domain containing-3 (NLRP3) inflammasome. Once activated, the NLRP3 inflammasome cleaves procaspase-1 to its mature form (caspase-1), which later cleaves IL-1β, IL-18, and gasdermin D. IL-1 and IL-18 are highly proinflammatory cytokines released into the extracellular space partly through transmembrane pores formed by gasdermin D108-109. All pathophysiological factors are summarized in Figure 1.
Liver fibrosis is the pathological characteristic that predicts the various results associated with liver injury in fatty liver110,111 and its genesis, liver fibrogenesis, or scar formation. It is a dynamic process of extracellular matrix (ECM) accumulation in which the hepatic stellate cell (formerly known as lipocyte, Ito cell, or perisinusoidal cell) is its primary source in the normal and fibrotic livers112.
Chronic liver damage paracrinely stimulates neighboring cells, such as sinusoidal endothelial cells, Kupffer cells, monocytes, and platelets, among others, producing an angiogenic stimulus with the formation of new blood vessels, sinusoidal remodeling and the release of multiple mediators such as platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and related receptors, and vasoactive mediators including nitric oxide and carbon monoxide113. All this together activates the stellate cells that increase the production of collagens (types I, III, and IV), several glycoproteins (cellular fibronectin, laminin, osteonectin, tenascin, von Willebrand factor), proteoglycans and glycosaminoglycans (perlecan, decorin, among others). It leads to a progressive accumulation and compositional change of the ECM that activates positive feedback pathways and further amplifies fibrosis through processes involving cell membrane receptors and other cytokines and adhesion proteins. One of these well-characterized pathways is that of integrins, a family of membrane proteins that control various cell functions, including gene expression, cell growth, and differentiation114.
For its part, fibrosis reflects a balance between the production and degradation of the ECM, a delicate balance between calcium-dependent enzymes that specifically degrade collagens and non-collagenous substrates, known as matrix metalloproteinases (MMPs or matrixins), and related specific inhibitors, known as tissue inhibitors of metalloproteinases (TIMPs). The substitution of the low-density matrix for that of the interstitial type, characteristic of fibrosis, has consequences on the function of hepatocytes, hepatic stellate cells, and endothelial cells, which partly explains the synthetic and metabolic dysfunction observed in patients with advanced fibrosis112-114.
Natural history
Paired biopsy studies showed that the natural history of fatty liver is very dynamic: patients with simple steatosis have a low risk of progression to cirrhosis, while in patients with NASH, this risk is increased; however, the process can be reversible, and some people will have spontaneous improvement16,115-120. Fibrosis seems to be the determinant of overall mortality and the outcomes associated with liver disease113,114,121,122. The damage caused by NAFLD can take many years to progress and is most often described in five stages, from 0 to 4, depending on the amount and distribution pattern of fibrosis found, similar to the METAVIR classification (F0-F4)123.
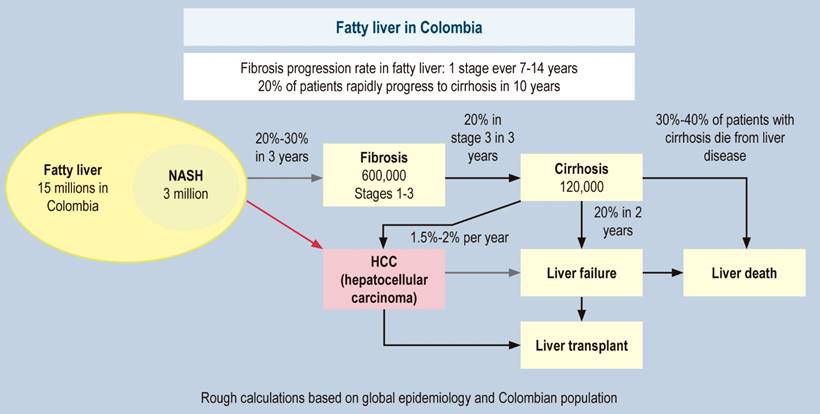
In fatty liver, considering that the data are average figures and non-linear, in all patients, fibrosis worsens by one stage every 14 years, while in NASH, it worsens by one stage every seven years120. Previous studies conclude that approximately 20% of the cases of simple steatosis progress to NASH; of these, approximately 20% progress to cirrhosis, with hepatocellular carcinoma (HCC) in 5% to 10% of them120,124-126. Recent analyses showed that 20% of patients with F3 progressed to cirrhosis in two years; of these, 20% of patients with compensated cirrhosis decompensated127,128. These figures are called the 20% “rule” and corroborate the previous data. In some cases, the liver can be damaged much faster than these average numbers, and one in five patients with fibrosis are rapid progressors120; this could be due to fluctuations in the severity of metabolic risk factors, the impact of various (unhealthy) lifestyles, and genetic factors. NASH is now recognized as the leading cause of cryptogenic cirrhosis (Figure 2)129. Since in Colombia, there are no exact data on our natural history, Figure 3 presents a forecast of the disease for our country based on international data.
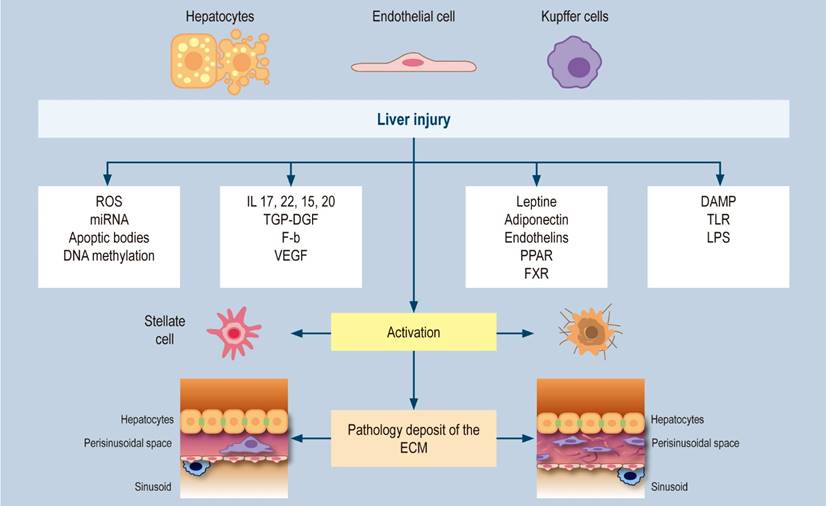
Figure 2 Hepatic fibrogenesis. Modified from: Tsuchida T et al. 2017;14(7):397-411; Friedman SL. Nat Rev Gastroenterol Hepatol. 2010;7(8):425-36; Kisseleva T et al. Nat Rev Gastroenterol Hepatol. 2021;18(3):151-166.