











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
Permalink
Introduction
Sjögren’s syndrome (SS) is the second most common autoimmune rheumatologic disease. It can manifest as a primary disorder (primary SS). Still, approximately one-third of patients have another underlying autoimmune condition (secondary SS), such as rheumatoid arthritis, systemic lupus erythematosus, scleroderma, or hypothyroidism. The key signs are xerostomia and xerophthalmia, although it can also involve the entire gastrointestinal tract1. It is diagnosed predominantly in middle-aged women (40-55 years) and is increasingly found in all races2. About a third of patients present with extra glandular manifestations, and 5% may develop a hematological malignancy. Genetic and environmental factors participate in its physiopathogenesis3.
Any part of the digestive system can be affected, most commonly in the form of dysmotility, gastroesophageal reflux disease (GERD), or atrophic gastritis4. Between 5% and 10% of patients with SS may have some degree of liver involvement and usually present with elevated liver enzymes in a subclinical and asymptomatic manner5. In these cases, liver biopsy may demonstrate characteristics of primary biliary cholangitis (PBC), autoimmune hepatitis (AIH), or primary sclerosing cholangitis (PSC), which is why they are serious complications that should be suspected in patients with altered liver function tests and autoantibodies with confirmatory findings in the liver biopsy6.
There are few cases in the literature about SS and PSC7 in which common characteristics of the condition have been demonstrated, such as abdominal pain, jaundice, and diarrhea, associated with other pathologies such as chronic pancreatitis and autoimmune processes such as retroperitoneal fibrosis, which favor clinical suspicion. Therefore, it is vital to investigate immunoglobulin G4 (IgG4)-related diseases, such as autoimmune pancreatitis or retroperitoneal fibrosis6. Below, on the one hand, we present the case of a patient with SS who had subclinical-asymptomatic hepatobiliary involvement, initially with altered liver function tests with a cholestatic pattern and presence of autoantibodies, and finally showed a pattern of liver disease compatible with PSC, in whom IgG4 levels were normal. On the other hand, the spectrum of gastrointestinal compromise in SS is briefly reviewed.
Case presentation
We present the case of a 70-year-old female patient who, 15 years ago, had dry ocular and oral syndrome associated with xeroderma, with arthralgias of mixed characteristics. She was evaluated by rheumatology, who requested an immunological profile with findings of positive antinuclear antibodies (ANA) with a 1:320 speckled pattern, positive anti-Ro and anti-La antibodies (qualitative test, fluorescence enzyme immunoassay), and SS-compatible salivary gland biopsy.
Pilocarpine was started with the improvement of dry oral symptoms and topical ocular treatment by ophthalmology. Five years ago, she was referred to hepatology due to aspartate aminotransferase (AST) 77 mg/dL, alanine aminotransferase (ALT) 56 mg/dL, alkaline phosphatase 960 mg/dL, γ-glutamyl transferase (GGT: 632 mg/dL, and total bilirubin 1.3 mg/dL. She had no previous history of consumption of hepatotoxic drugs, nor history of hepatic steatosis associated with metabolic dysfunction, nor of chronic cardiopulmonary diseases. The viral profile for hepatitis B and hepatitis C viruses was negative. Subsequently, antimitochondrial antibodies (AMA) and IgG4 were normal. Antineutrophil cytoplasmic antibodies (p-ANCA) were not performed. Imaging studies such as magnetic resonance cholangiography (MRC) were performed with findings of loss of the caudate lobe, segmental dilation of the intrahepatic bile duct, and a beaded appearance associated with the common bile duct dilation. These findings were seen in the context of PSC (Figure 1).

Figure 1 MRC with findings of a beaded pattern of the intrahepatic bile duct and dilation of the common bile duct. Image owned by the authors.
She has received treatment with ursodeoxycholic acid (UDCA) 900 mg/day (recommended at a dose of up to 15 mg/kg/day)8,9 with partial improvement of the cholestatic pattern. After the diagnosis and during the evolution of her disease, the patient has had three episodes of cholangitis in which endoscopic retrograde cholangiopancreatography (ERCP) has been performed without findings of stones, with little bile sludge and purulent bile output, precipitated by her underlying liver disease.
Gastrointestinal involvement in sjögren’s syndrome
In addition to the classic manifestation of xerostomia and xerophthalmia, another set of symptoms related to the gastrointestinal tract and liver may occur in SS. These manifestations can typically be related either to a decrease in saliva production due to the involvement of the exocrine glands or secondary to the autoimmune process that allows glandular damage5. The main alterations are described below according to the affected gastrointestinal organ (Figure 2).
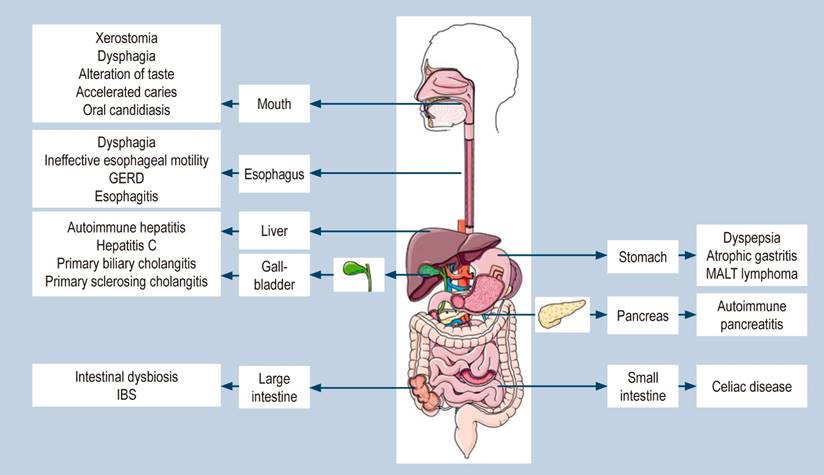
Figure 2 Gastrointestinal manifestations associated with SS. Diagram made using images from Elsevier Medical Art, under its conditions of use. Own work based on1-5.
Mouth
A lymphocytic inflammatory infiltrate in the salivary and lacrimal glands destroys glandular tissue, and the most common initial symptoms include xerostomia and xerophthalmia. The spectrum of its manifestations is broad and ranges from sensitivity to acidic and spicy foods, dysphagia, dysgeusia, and, in the most severe cases, accelerated caries, tooth loss, hoarseness, and oral candidiasis, among others5. The oral mucosa is opaque, parchment-like, and adheres to the examining finger. Angular cheilitis, reduced infralingual salivation, and enlarged parotid or submandibular glands may exist. Saliva cannot be squeezed out by massaging the salivary glands10. There may be alterations in the neurotransmission of the parasympathetic nervous system, leading to glandular dysfunction and interference in excitatory stimuli from the vagal and sacral nerves to the gastrointestinal tract11. Treatment of oral symptoms includes topical agents and secretagogues of muscarinic receptor stimulants (pilocarpine and cevimeline). Pilocarpine is a muscarinic cholinergic agonist and binds to muscarinic M3 receptors, and from clinical trials, it is known to increase salivary flow and improve oral symptoms12.
Esophagus
Dysphagia is a frequent problem secondary to a decrease in saliva production or alteration in motility. It occurs in up to a third of patients. It is secondary to inflammatory infiltrate in the exocrine glands of the esophagus, which affects the esophageal muscles with subsequent deterioration in the motor coordination of swallowing13.
It should be highlighted that the average saliva production from the salivary gland is the critical component in the swallowing mechanism. Dysphagia is relatively common in SS and is usually related to the absence of saliva. However, among the earliest signs of dry mouth is difficulty starting the swallowing process, called pharyngeal dysphagia. One of the standard swallowing mechanisms begins in the throat, and both esophageal lubrication and adequate coordination of esophageal contractions are necessary5.
In addition to the decrease in saliva production by the secretory glands, the mucus-producing cells of the esophagus may also be affected, causing esophageal dryness and the sensation of difficult passage of food through the esophagus. Abnormalities of parasympathetic neurotransmission may contribute to glandular dysfunction in SS14.
In one study, IgG antibodies against muscarinic receptors were described in the serum of patients with SS, possibly interfering with excitatory stimuli to the vagal and sacral nerves of the gastrointestinal tract. However, in another study, these antibodies were not detected11,15.
Difficulty swallowing can also indicate a lack of coordination of the contraction waves that allow food to pass through the esophagus, known as esophageal dysmotility; this involvement can be as frequent as in other connective tissue diseases, such as scleroderma. It can appear in up to a third of patients with SS and cause dysphagia in 30-80% of them16.
The evaluation of patients with difficulty swallowing typically includes an esophagogastroduodenoscopy (EGD) and esophagram to assess the anatomy of the gastrointestinal tract. In these studies, esophageal dysmotility may be suspected, although it is necessary to complement them with esophageal manometry to evaluate the motor component more accurately. Research in which esophageal manometry was performed on patients with SS has described normal studies and alterations such as low pressures of the upper esophageal sphincter17, aperistalsis, non-peristaltic tertiary contractions, ineffective esophageal motility, achalasia17,18, and decreased pressure of the lower esophageal sphincter19.
Other studies have observed no correlation between dysphagia and esophageal dysmotility16. A study on a cohort of Colombian patients with esophageal manometric alterations in connective tissue diseases describes 21 patients according to the American College of Rheumatology (ACR) classification criteria, of which 52.3% had SS. Most of them had ineffective esophageal motility20.
GERD is ubiquitous, affecting up to 20% of the population and producing symptoms ranging from chest pain to epigastralgia5. Prolonged gastroesophageal reflux can cause esophageal lesions, the most common being esophagitis and subsequent metaplastic changes (Barrett’s esophagus), resulting in potentially malignant lesions. Laryngoesophageal reflux can also produce other symptoms, such as halitosis, chronic cough, pharyngeal dysphagia, and a foreign body sensation in the throat13. Salivary flow usually sweeps away the contents of gastroesophageal reflux and serves as a buffer in the presence of gastric acid reflux21. Reduced salivary flow and altered coordination of esophageal contractions are essential factors that contribute to developing GERD and laryngopharyngeal reflux in SS5.
GERD is initially treated with lifestyle changes, including weight loss, elevation of the head of the bed, cessation of tobacco use, and avoidance of excessive alcohol consumption. Additionally, eating large amounts of food two hours before bedtime should be avoided. If these measures are insufficient, treatment should be initiated with medications that reduce acid production, such as H2 blockers and proton pump inhibitors (PPIs)5.
Stomach
Among the upper gastrointestinal manifestations in the stomach, nausea, epigastric discomfort, heartburn, and dyspepsia stand out in up to 15-23%22, which are relatively common in patients with SS.
Symptoms are commonly related to GERD and typically initiate PPI medication. However, some patients with SS have antibodies against parietal cells that cause hypochlorhydria, atrophic gastritis5, and vitamin B12 malabsorption due to a lack of intrinsic factors23,24.
Vitamin B12 is vital for producing red blood cells, and when there is a deficiency, pernicious anemia or peripheral neuropathy occurs, which can be easily managed with timely replacement with intramuscular cyanocobalamin. However, this finding is not very common in patients with SS. Atrophic gastritis is diagnosed directly by biopsy of the gastric wall during endoscopy. In patients undergoing endoscopy, chronic atrophic gastritis was found in 25% to 81%24. In agreement with this finding, hypopepsinogenemia was found in 69% of patients, half with elevated serum gastrin25.
Chronic inflammation or glandular atrophy studied by immunohistochemistry revealed a predominance of CD3+ T lymphocytes, mostly CD4+23. These findings are similar to those found in the salivary glands, further supporting SS as a systemic disease. Although basal acid production was decreased in most patients, it was lower in patients with body-localized type gastritis with elevated serum gastrin levels. Some patients with dyspeptic symptoms should be evaluated for Helicobacter pylori by respiratory testing (urease testing) or direct biopsy. Detecting antigens in fecal matter is very useful but is not done routinely. Blood antibodies are not routinely recommended. SS is associated with increased MALT (mucosa-associated lymphoid tissue) lymphoma, one of the absolute indications for eradicating H. pylori. In turn, it must be considered that in these patients, the response rate is lower than in the general population5,22,26.
Small intestine
In the small intestine, celiac disease, also known as gluten enteropathy, is a common condition in which the immune system responds abnormally to gluten, a protein found in some foods, causing damage to the small intestinal wall5,27. Celiac disease may affect up to one in 300 individuals but may be up to ten times more prevalent in patients with SS than in the general population. The increased prevalence of the celiac disease has been seen in other conditions mediated by the immune system, such as autoimmune thyroiditis or type 1 diabetes. Symptoms of celiac disease include diarrhea, constipation, fatigue, weight loss, flatulence, and abdominal discomfort, conditions associated with nutrient and vitamin deficiencies that frequently result in osteoporosis and anemia.
The diagnosis of celiac disease is difficult if based on symptoms alone, which is why gastrointestinal autoantibody tests should be performed and requested in patients with gastrointestinal symptoms or nutritional deficiencies. EGD can demonstrate villous alterations and histological changes; gluten elimination typically allows the resolution of small intestine damage28,29. Among other intestinal compromises, although very rare, intestinal infarction or intestinal ischemia due to vasculitis may occur when SS is associated with cryoglobulinemia30.
Colon
The involvement of the colon as an extra glandular manifestation of SS has not been well characterized, and it is still not clear to what degree it can affect it. Constipation and diarrhea can occur frequently, and their association with irritable bowel syndrome (IBS)27, inflammatory bowel disease (IBD)31, diverticular disease, colonic adenomas, malignant tumors26, and, more recently, intestinal dysbiosis has been described32. A large proportion of patients with SS have the typical symptoms of IBS or dysmotility. It has also been outlined that the dry complex may represent an extraintestinal manifestation of IBS33, and there are doubts about the actual association of IBS with SS. Diverticular disease has no direct relationship with SS; however, we cannot exclude the possibility that they occur more frequently in SS, favored by constipation, due, on the one hand, to the disease itself and, on the other hand, to anti-inflammatory drugs27.
It is also known that intestinal dysbiosis is a prevalent problem in SS, which exceeds 20% of cases34. Intestinal dysbiosis is favored by the compromised integrity of the epithelium and its barrier function in SS, possibly leading to a disturbed microbiota-host immune system interaction. It may reduce and amplify local and systemic inflammatory disease35. Low levels of bacteria from the genera Alistipes and Bifidobacterium and a moderate correlation of serum levels of fecal calprotectin with disease activity have even been reported; if >150 μg/g, it may indicate a concomitant organic disease (for example, neoplasia or IBD) and warrants additional studies34.
Pancreas
Pancreatitis may result from the same autoimmune inflammation that affects the salivary and lacrimal glands. Autoimmune pancreatitis is a rare complication in SS. It is characterized by diffuse inflammation of the pancreas and narrowing of the main pancreatic duct, which results in abdominal pain, nausea, and emesis. This condition has been described as primary pancreatitis and may be associated with another autoimmune condition. Elevation of IgG4 levels helps confirm the diagnosis of autoimmune pancreatitis type 1. The correlation between elevated IgG4 levels and patients with SS may currently be a systemic disease associated with sclerosing sialadenitis, PSC, retroperitoneal fibrosis, or non-infectious aortitis. Early diagnosis can prevent the development of chronic pancreatitis and pancreatic insufficiency36,37.
Hepatobiliary system
Up to 7% of patients with SS may have some degree of liver involvement, usually with elevated liver enzymes that occur subclinically and asymptomatically. In these patients, liver biopsies demonstrate characteristics of PBC, AIH, or PSC5,38.
PBC is an autoimmune liver disease associated with SS, in which the small bile ducts are attacked and destroyed by the immune system. Continued inflammation and damage to these bile ducts allow for scarring and the production of cirrhosis and liver failure. In SS, the elevation of liver function tests and the detection of AMAs suggest the diagnosis of PBC; however, a biopsy is generally necessary to confirm this involvement39.
Approximately 40-50% of patients with PBC will have dry symptoms that mimic SS, including xerophthalmia and xerostomia7,40. Skin itching is one of the most common symptoms of PBC, which, in mild cases, can be managed with emollients. In contrast, adding non-absorbent resins such as cholestyramine or colestipol can provide itch control in severe cases. These resins bind many substances in the gastrointestinal tract and inhibit the reabsorption of bile salts5.
Other symptoms of PBC complications include hyperpigmentation, arthritis, metabolic bone disease (osteoporosis and osteomalacia), and fat-soluble vitamin deficiency. In the late stages, when there is already cirrhosis, jaundice, edema, portal hypertension, and ascites are included5.
Regarding treatment, the only drug that can modify the natural history and progression of PBC is UDCA41. When there is a therapeutic failure and disease progression, liver transplantation is considered39,42.
AIH is characterized by chronic inflammation that can eventually lead to cirrhosis. The majority of patients with AIH present with nonspecific symptoms such as fatigue, lethargy, hyporexia, and arthralgia; rarely, AIH causes acute liver failure. AIH is suspected in patients with SS who develop a progressive increase in liver enzymes without another explanation, such as due to medications. This entity is diagnosed with a combination of autoantibodies and specific characteristics of the liver biopsy. Not all patients with AIH need treatment; the decision to start it depends on the severity of the disease and includes glucocorticoids and immunomodulators such as azathioprine40.
PSC is a chronic cholestatic liver disease characterized by structural alterations of both the intrahepatic and extrahepatic bile ducts, in which obliterative fibrosis prevails. It is an autoimmune pathology frequently associated with positive autoantibodies, including ANA and p-ANCA. Few cases reported in the literature have described SS and PSC, in which common characteristics of the manifestation of the condition have been found, such as abdominal pain, jaundice, and diarrhea7. It is associated with other pathologies, such as chronic pancreatitis, and autoimmune processes, such as retroperitoneal fibrosis, which can favor clinical suspicion and, in turn, an early diagnosis of PSC in patients with SS. It is also important to inquire about other diseases related to IgG4 in patients with the coexistence of these diseases, such as autoimmune pancreatitis or retroperitoneal fibrosis6.
Hepatitis C virus infection is not more prevalent in SS than in the general population. However, this infection can produce a syndrome similar to Sjögren’s, given by SS. Elevation of liver enzymes in hepatitis C infection produces xerostomia and xerophthalmia, arthralgia, and lymphocytic sialadenitis that mimics the classic Sjögren manifestation. Still, although the hepatitis C virus can produce ANA and positive rheumatoid factor, anti-Ro or anti-La antibodies do not become positive, differentiating these two pathological entities43,44.
Discussion
Some characteristics of the presented clinical picture draw attention and deserve to be analyzed in the light of current knowledge.
The patient was asymptomatic and presented with a cholestatic profile and recorded alkaline phosphatase values more than three times above normal without eosinophilia, so an MRC was performed that confirmed the diagnosis of PSC, while the IgG4 and AMA were negative. In PSC, around 50% of patients are asymptomatic at initial manifestation with a history of abnormal liver chemistry45. View of the biliary tree is the cornerstone of the diagnosis, and ERCP is the preferred method to establish it. Band stenosis and beaded formations have been described as highly specific for PSC. However, MRC is the first test of choice, as it is non-invasive and has diagnostic accuracy comparable to ERCP45, with a sensitivity of 86% and specificity of 94% for diagnosing PSC41,46. The evaluation of AMA will allow differentiation from PBC since, in CEP, it will be absent47. Measurement of serum IgG4 (a characteristic marker of autoimmune pancreatitis seen at lower levels in several other diseases) will allow the discernment of IgG4-associated cholangitis from PSC48, and it has been reported that up to 9% of patients may present values above the upper limit of normality49.
Although there is a strong association of PBC with SS, very few cases of SS associated with PSC have been described, and this association is considered sporadic. When associated, they usually present with chronic pancreatitis. Pancreatic hyposecretion is a characteristic finding in SS and may be anticipatory in PBC; however, it is unclear that it may exist in patients with SS associated with PSC50. It is worth mentioning that in PSC, there may be a diverse group of diseases and that different etiologies may be involved, resulting in a typical clinical picture51. In SS, at least one-third of patients develop extra glandular manifestations of the disease (beyond the salivary and lacrimal glands), which gives rise to a broad clinical spectrum due to the numerous possible systemic manifestations, serological findings, and complications14. In the patient in this case, SS, positive ANA with a speckled pattern, positive anti-Ro and anti-La antibodies, and compatible salivary gland biopsy were confirmed without findings for any other connective tissue disease. From the hepatobiliary point of view, the course was also asymptomatic, and at the time of the diagnosis of PSC, no symptoms were suggesting chronic pancreatitis.
In the pathogenesis of PSC, the hypotheses regarding the initial trigger include infection or another immunological stimulus in individuals with underlying genetic predisposition, immunological lesions, and vascular lesions, among others52. The pathophysiological process of inflammation and fibrotic processes can be ongoing for years before clinical appearance6,50,52. In SS, it has been proposed that in the pathogenesis of the systemic disease, most extra-glandular manifestations are considered an expression of autoimmune epithelitis since the epithelial component is the main target of the autoimmune response3,53. In the case presented, the association of SS and PSC suggests an underlying autoimmune mechanism, as it was a patient with generalized sicca manifestations with xerostomia and xerophthalmia (dry eye) and a concomitant disorder such as PSC.
Despite findings of a cholestatic liver profile, cholangiographic abnormalities, and high levels of circulating ANA, it progressed without pruritus, without high circulating levels of IgG or anti-smooth muscle antibodies, nor was ulcerative colitis (UC) documented throughout the disease, so it was not conclusive for overlapping. Initial treatment included UDCA, and there was no requirement for immunosuppression. In the study by Floreani et al.54, the clinical characteristics of seven patients with AIH-PSC overlap were compared with those of 34 patients with classic PSC, finding that patients with PSC-AIH overlap were of younger ages at manifestation (mean age 21 vs. 32 years), significantly higher aminotransferases, baseline clinical characteristics of AIH at presentation and subsequent diagnosis of PSC when immunosuppression did not achieve clinical remission, as well as prevalence of autoantibodies and levels of serum immunoglobulin. PSC is also more common in middle-aged patients with IBD, and men represent 60-70% of patients with UC and PSC41.
From three meta-analyses, it is known that UDCA at doses of up to 15 mg/kg/day has been associated with an improvement in liver biochemistry and stabilization of liver inflammation, but without influencing long-term clinical outcomes, which include survival and delay in the need for liver transplant8,9,55. Treatment with high doses (28 to 30 mg/kg per day) is associated with an increased risk of varicose veins, death, and liver transplantation in the following five years41,42. A 2010 guideline issued by the American Association for the Study of Liver Diseases (AASLD) advises against the use of UDCA for the specific treatment of PSC(56), while, in 2015, the American College of Gastroenterology (ACG) did not. There is no recommendation for UDCA other than it should not be used in doses higher than 28 mg/kg/day41. In this regard, in PSC, UDCA is used to control cholestasis. Although standard dosing of UDCA has not been associated with adverse outcomes, it cannot be routinely recommended until more data on efficacy and safety are available.
Given the documentation of biliary stricture in ERCP, endoscopic dilation with or without stent placement may be required41,45. Corticosteroids and other immunosuppressive agents are recommended in patients with overlap syndromes41. A Cochrane review with 13 treatments evaluated in 22 randomized trials with 2,211 participants did not demonstrate the efficacy of medical therapy in PSC57. A liver transplant is the only treatment shown to prolong survival42.
Conclusions
Any part of the gastrointestinal system can be affected by SS. The role of medications, infections, diet, nutrition, and tumor lesions must be considered.
The association of PSC and SS is rare; it is defined as having an autoimmune cause, and its treatment depends on the PSC component.
As this case shows, PSC can occur silently in SS, and simple procedures such as measuring the liver profile and subsequent CMR can lead to its identification.

