










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista colombiana de Gastroenterología
versão impressa ISSN 0120-9957versão On-line ISSN 2500-7440
Rev. colomb. Gastroenterol. vol.38 no.3 Bogotá jul./set. 2023 Epub 18-Jan-2024
https://doi.org/10.22516/25007440.918
Reporte de caso
Compromiso gastrointestinal y hepático en síndrome de Sjögren primario: reporte de caso y revisión de la literatura
1
 http://orcid.org/0000-0003-2361-1018
http://orcid.org/0000-0003-2361-1018
2
*
http://orcid.org/0000-0003-2735-2922
3
http://orcid.org/0000-0001-8626-1567
3
http://orcid.org/0000-0001-5714-1953
1Internist, gastroenterologist, rheumatologist, Hospital Internacional de Colombia. Bucaramanga, Colombia.
2Gastroenterology fellow. Universidad Nacional de Colombia. Bogotá, Colombia.
3Internist, gastroenterologist, Hospital Universitario San Ignacio. Bogotá, Colombia.
4Internist, gastroenterologist, epidemiologist, Hospital Internacional de Colombia, Gastroadvanced IPS. Bucaramanga, Colombia.
El síndrome de Sjögren es una enfermedad autoinmune sistémica que se caracteriza por la sequedad ocular y bucal debido a la afección de glándulas exocrinas; sin embargo, puede manifestarse con síntomas gastrointestinales que abarcan un espectro amplio desde la dismotilidad esofágica e intestinal, acalasia, hipoclorhidria y gastritis crónica atrófica hasta enzimodeficiencia pancreática, disfunción biliar y cirrosis hepática, que tiene variación en sus manifestaciones clínicas y se asocia con abordajes erróneos en muchas ocasiones. En este artículo se hace una revisión acerca de las manifestaciones gastrointestinales de síndrome de Sjögren y se presenta el caso de una mujer en la octava década de la vida con este síndrome, que cursa con enfermedad hepatobiliar asintomática, documentación de alteración en pruebas de perfil hepático y diagnóstico ulterior de colangitis esclerosante primaria, por lo que recibió un tratamiento inicial con ácido ursodesoxicólico. Durante el curso de su enfermedad ha presentado 3 episodios de colangitis, con requerimiento de colangiopancreatografía retrógrada endoscópica sin hallazgos de cálculos, con escaso barro biliar y salida de bilis purulenta, precipitada por su enfermedad hepática de base. La asociación entre el síndrome de Sjögren y la colangitis esclerosante primaria es infrecuente y justifica una consideración especial.
Palabras clave: Síndrome de Sjögren; colangitis esclerosante; anomalías del sistema digestivo; anticuerpos antinucleares; enfermedades gastrointestinales
Sjögren’s syndrome is a systemic autoimmune disease characterized by dry eyes and mouth due to the involvement of exocrine glands. However, it can manifest with GI symptoms that cover a broad spectrum from esophageal and intestinal dysmotility, achalasia, hypochlorhydria, and chronic atrophic gastritis to pancreatic enzyme deficiency, biliary dysfunction, and liver cirrhosis, which varies in its clinical manifestations and is often associated with erroneous approaches. This article reviews the GI manifestations of Sjögren’s syndrome. It presents the case of a woman in her eighth decade of life with this syndrome. She showed asymptomatic hepatobiliary disease, documented abnormalities in liver profile tests, and a subsequent diagnosis of primary sclerosing cholangitis, for which she received initial treatment with ursodeoxycholic acid. During her condition, the patient has had three episodes of cholangitis, requiring endoscopic retrograde cholangiopancreatography with no findings of stones, with scant biliary sludge and discharge of purulent bile precipitated by her underlying liver disease. The association between Sjögren’s syndrome and primary sclerosing cholangitis is rare and calls for special consideration.
Keywords: Sjögren’s syndrome; sclerosing cholangitis; digestive system abnormalities; antinuclear antibodies; gastrointestinal diseases
Introducción
El síndrome de Sjögren (SS) es la segunda enfermedad reumatológica autoinmune más frecuente, puede presentarse como un trastorno primario (síndrome de Sjögren primario), pero aproximadamente un tercio de los pacientes tiene otra afección autoinmune subyacente (síndrome de Sjögren secundario), como artritis reumatoide, lupus eritematoso sistémico, esclerodermia o hipotiroidismo. Los signos claves son xerostomía y xeroftalmia, aunque también puede comprometer la totalidad del tracto gastrointestinal1. Se diagnóstica predominantemente en mujeres, en la mediana edad (40-55 años) y cada vez se encuentra más en todas las razas2. Alrededor de un tercio de los pacientes se presenta con manifestaciones extraglandulares y el 5% puede desarrollar una neoplasia hematológica. En su fisiopatogenia participan factores genéticos y ambientales3.
Cualquier parte del sistema digestivo puede verse afectada, y más comúnmente se encuentra dismotilidad, enfermedad por reflujo gastroesofágico (ERGE) o gastritis atrófica4. Entre 5% y 10% de los pacientes con SS pueden tener algún grado de compromiso hepático, y cursan de modo usual con elevación de enzimas hepáticas de manera subclínica y asintomática5. En estos casos, la biopsia hepática puede demostrar características de colangitis biliar primaria (CBP), hepatitis autoinmune (HAI) o colangitis esclerosante primaria (CEP), por lo que son complicaciones serias que deben ser sospechadas en pacientes con pruebas de función hepática alteradas y presencia de autoanticuerpos con hallazgos confirmatorios en biopsia hepática6.
Existen pocos casos reportados en la literatura acerca de la presencia de SS y CEP7, en los que se han demostrado características comunes de la presentación del cuadro, como dolor abdominal, ictericia y diarrea, asociándose a presencia de otras patologías como pancreatitis crónica y procesos autoinmunes como la fibrosis retroperitoneal, que favorecen la sospecha clínica, por lo que es importante indagar sobre enfermedades relacionadas con inmunoglobulina G4 (IgG4), como pancreatitis autoinmune o fibrosis retroperitoneal6. A continuación, por una parte, se presenta el caso de una paciente con SS quien cursó con compromiso hepatobiliar de modo subclínico-asintomático, inicialmente con pruebas de función hepática alteradas con patrón colestásico y presencia de autoanticuerpos, y finalmente presentó un patrón de enfermedad hepática compatible con CEP, en quien los niveles de IgG4 fueron normales. Por otra parte, se revisa brevemente el espectro del compromiso digestivo en SS.
Presentación del caso
Se trata de una paciente femenina de 70 años que hace 15 años presentó un síndrome seco ocular y oral, asociado a xerodermia, con artralgias de características mixtas. Fue evaluada por reumatología, que solicitó un perfil inmunológico con hallazgos de anticuerpos antinucleares (ANA) positivos con patrón moteado 1:320, con anticuerpos anti-Ro y anti-La positivos (prueba cualitativa, inmunoensayo enzimático de fluorescencia) y biopsia de glándula salival compatible con SS.
Se inició la pilocarpina con mejoría de los síntomas secos orales y tratamiento tópico ocular por parte de oftalmología. Desde hace 5 años fue remitida a hepatología por presencia de aspartato-aminotransferasa (AST) 77 mg/dL, alanina-aminotransferasa (ALT) 56 mg/dL, fosfatasa alcalina de 960 mg/dL, γ-glutamil transferasa (GGT): 632 mg/dL y bilirrubina total de 1,3 mg/dL. No tenía antecedente previo de consumo de medicamentos hepatotóxicos, ni historia de esteatosis hepática asociada a disfunción metabólica, ni de enfermedades cardiopulmonares crónicas. El perfil viral para virus de hepatitis B y hepatitis C fue negativo. Ulteriormente, los anticuerpos antimitocondriales (AMA) y la IgG4 fueron normales. No se realizaron anticuerpos anticitoplasma del neutrófilo (p-ANCA). Se realizaron estudios imagenológicos como la colangiorresonancia magnética (CRM) con hallazgos de pérdida del lóbulo caudado, con dilatación segmentaria de la vía biliar intrahepática con aspecto arrosariado asociado a dilatación del colédoco, hallazgos que fueron vistos en el contexto de una CEP (Figura 1).
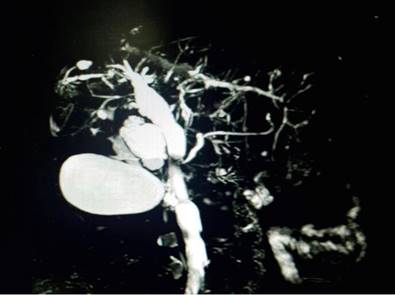
Figura 1 CRM con hallazgos de patrón arrosariado de la vía biliar intrahepática y dilatación del colédoco. Imagen propiedad de los autores.
Ha recibido tratamiento con ácido ursodesoxicólico (UDCA) 900 mg/día (recomendado a una dosis de hasta 15 mg/kg/día)8,9 con mejoría parcial del patrón colestásico. Después del diagnóstico, durante la evolución de su enfermedad la paciente ha presentado 3 episodios de colangitis en los cuales se han realizado colangiopancreatografía retrógrada endoscópica (CPRE) sin hallazgos de cálculos, con escaso barro biliar y salida de bilis purulenta, precipitada por su enfermedad hepática de base.
Compromiso digestivo en síndrome de sjögren
Adicionalmente a la clásica presentación de xerostomía y xeroftalmia, en el SS puede presentarse otro conjunto de síntomas relacionados con el tracto gastrointestinal y el hígado. Estas manifestaciones pueden estar típicamente relacionadas ya sea con la disminución de la producción de saliva por el compromiso de las glándulas exocrinas o secundario al proceso autoinmune que permite el daño glandular5. A continuación se describen las principales alteraciones según el órgano gastrointestinal afectado (Figura 2).
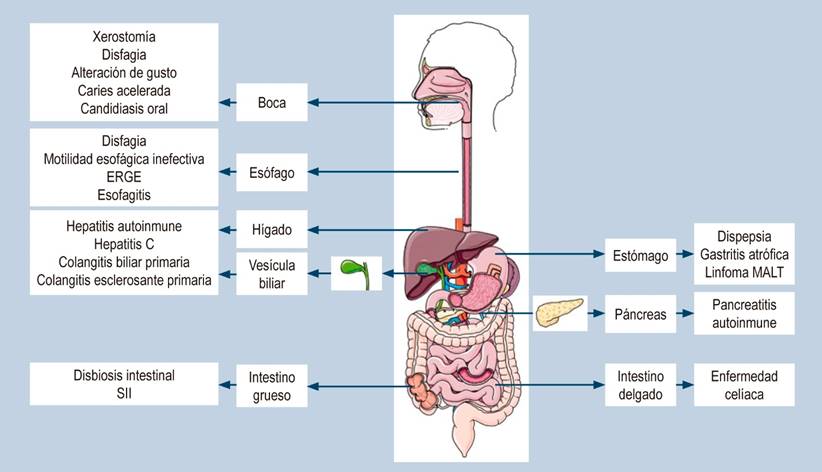
Figura 2 Manifestaciones gastrointestinales asociadas a SS. Esquema realizado utilizando imágenes de Elsevier Medical Art, bajo sus condiciones propias de uso. Elaboración propia con base en1-5.
Boca
La presencia de un infiltrado inflamatorio linfocitario en las glándulas salivales y lagrimales provoca destrucción del tejido glandular y los síntomas más comunes iniciales incluyen xerostomía y xeroftalmia. El espectro en sus manifestaciones es amplio y va desde sensibilidad a los alimentos ácidos y picantes, disfagia, disgeusia y, en los casos más graves, se pueden presentar caries aceleradas, pérdidas dentales, ronquera y candidiasis oral, entre otras5. La mucosa oral es opaca, parecida al pergamino y se adhiere al dedo examinador. Puede haber queilitis angular, reducción de la salivación infralingual y aumento del tamaño de la glándula parótida o submandibular. La saliva no puede ser exprimida ante el masaje de las glándulas salivales10. Puede haber alteraciones en la neurotransmisión del sistema nervioso parasimpático, lo que lleva a disfunción glandular e interferencia en estímulos excitatorios de los nervios vagales y sacros al tracto gastrointestinal11. El tratamiento de los síntomas orales incluye agentes tópicos y secretagogos, estimulantes de receptores muscarínicos (pilocarpina y cevimelina). La pilocarpina es un agonista colinérgico muscarínico y se une a los receptores muscarínicos M3, y a partir de ensayos clínicos se sabe que aumenta el flujo salival y mejora los síntomas orales12.
Esófago
La disfagia es un problema frecuente que puede ser secundario a la disminución en la producción de saliva o a la alteración en la motilidad que se presenta hasta en un tercio de los pacientes y es secundario a la presencia de infiltrado inflamatorio en las glándulas exocrinas del esófago, lo que afecta la musculatura esofágica con el posterior deterioro en la coordinación motora de la deglución13.
En el esófago es importante resaltar que la producción normal de saliva de la glándula salival es el componente clave en el mecanismo de la deglución. La presencia de disfagia es relativamente común en el SS y usualmente se relaciona con ausencia de saliva; sin embargo, dentro de los signos más tempranos asociados a boca seca se encuentra la dificultad para iniciar el proceso de la deglución, llamado disfagia faríngea. Uno de los mecanismos normales de la deglución inicia en la garganta y son necesarias tanto la lubricación esofágica como la adecuada coordinación de las contracciones esofágicas5.
Adicionalmente a la disminución de la producción de saliva por las glándulas secretoras, las células productoras de moco del esófago también pueden estar afectadas, lo que también permite la sequedad esofágica y la sensación de difícil paso del alimento a través del esófago. Anormalidades de la neurotransmisión parasimpática pueden contribuir a la disfunción glandular en SS14.
En un estudio se describieron anticuerpos IgG contra los receptores muscarínicos en el suero de pacientes con SS, que posiblemente interfieren con los estímulos excitatorios de los nervios vagales y los nervios sacros del tracto gastrointestinal. Sin embargo, en otro estudio no se detectaron estos anticuerpos11,15.
La dificultad en la deglución también puede ser un signo de falta de coordinación de las ondas de contracción que permiten el paso de la comida a través del esófago, conocido como dismotilidad esofágica; este compromiso puede ser tan frecuente como en otras enfermedades del tejido conectivo, como en la esclerodermia, puede llegar a aparecer hasta en un tercio de los pacientes con SS y producir disfagia en el 30%-80% de ellos16.
Dentro de la evaluación de los pacientes con dificultad para la deglución típicamente se incluye una endoscopia de vías digestivas altas (EVDA) y esofagograma para evaluar la anatomía del tracto gastrointestinal; en estos estudios puede sospecharse dismotilidad esofágica, aunque es necesario complementarlos con manometría esofágica para evaluar el componente motor con mayor precisión. En estudios en los que se les realizó manometría esofágica a pacientes con SS se han descrito estudios normales y alteraciones desde presiones bajas del esfínter esofágico superior17, aperistalsis, contracciones terciarias no peristálticas, motilidad esofágica inefectiva, acalasia17,18 y disminución de la presión del esfínter esofágico inferior19.
Otros estudios han observado que no existe una correlación en la disfagia y la dismotilidad esofágica16. Un estudio realizado de una cohorte de pacientes colombianos, con alteraciones manométricas esofágicas en enfermedades de tejido conectivo, describe a 21 pacientes según los criterios clasificatorios del Colegio Estadounidense de Reumatología (ACR), de los cuales el 52,3% tenía SS, quienes en su mayoría presentaron motilidad esofágica inefectiva20.
La ERGE es muy común, afecta hasta el 20% de la población y produce sintomatología que va desde dolor torácico hasta epigastralgia5. La presencia de reflujo gastroesofágico prolongado puede producir lesiones esofágicas, las más comunes son la esofagitis y posteriores cambios metaplásicos (esófago de Barrett), que pueden condicionar lesiones potencialmente malignas. El reflujo laringoesofágico también puede producir otros síntomas como halitosis, tos crónica, disfagia faríngea y sensación de cuerpo extraño en la garganta13. El flujo salival normalmente barre el contenido del reflujo gastroesofágico y sirve como amortiguador ante la presencia de reflujo gástrico ácido21. La reducción del flujo salival y la alteración en la coordinación de las contracciones esofágicas son factores importantes que contribuyen al desarrollo de ERGE y reflujo laringofaríngeo en SS5.
La ERGE inicialmente se trata con cambios en el estilo de vida, que incluyen la pérdida de peso, elevación de la cabecera de la cama, suspensión del consumo de tabaco y evitación del consumo excesivo de alcohol; adicionalmente, se deben evitar las grandes cantidades de alimentos y su ingestión 2 horas antes de dormir. Si estas medidas no son suficientes, se debe iniciar el tratamiento con medicamentos que disminuyan la producción de ácido, como los bloqueantes H2 y los inhibidores de la bomba de protones (IBP)5.
Estómago
Dentro de las manifestaciones gastrointestinales altas en el estómago se destacan la presencia de náuseas, malestar epigástrico, pirosis y dispepsia hasta en un 15%-23%22, las cuales son relativamente comunes en los pacientes con SS.
Comúnmente, los síntomas se relacionan con la ERGE y típicamente inician medicación con IBP; sin embargo, algunos pacientes con SS tienen presencia de anticuerpos contra las células parietales que provocan hipoclorhidria, gastritis atrófica5 y malabsorción de vitamina B12, debido a la falta de factor intrínseco23,24.
La vitamina B12 es importante para la producción de glóbulos rojos y cuando hay deficiencia de esta se presenta anemia perniciosa o neuropatía periférica, la cual puede manejarse de manera sencilla con suplencia oportuna con cianocobalamina intramuscular, aunque no es muy común este hallazgo en pacientes con SS. El diagnóstico de gastritis atrófica se realiza directamente por biopsia de la pared gástrica durante la endoscopia. En los pacientes sometidos a endoscopia, la gastritis atrófica crónica se encontró en el 25% a 81% de los pacientes24. De acuerdo con este hallazgo, la hipopepsinogenemia se encontró en el 69% de los pacientes, la mitad con una gastrina sérica elevada25.
La inflamación crónica o atrofia glandular estudiada por inmunohistoquímica reveló un predominio de linfocitos T CD3+, en su mayoría CD4+23. Estos hallazgos son similares a los encontrados en las glándulas salivales, lo que sustenta aún más que el SS es una enfermedad sistémica. Aunque la producción de ácido basal disminuyó en la mayoría de los pacientes, fue menor en los pacientes con gastritis de tipo A localizada en el cuerpo con niveles elevados de gastrina sérica. Algunos pacientes con síntomas dispépticos deben ser evaluados para Helicobacter pylori por medio de pruebas respiratorias (prueba de ureasa) o por biopsia directa; la detección de antígenos en la materia fecal es de gran utilidad, pero no se hacen de rutina. Los anticuerpos en sangre no se recomiendan de rutina. El SS se asocia con un incremento de linfoma tipo MALT (tejido linfoide asociado a mucosas), el cual es una de las indicaciones absolutas de erradicación del H. pylori; a su vez, debe tenerse en cuenta que en estos pacientes la tasa de respuesta es menor que en la población general5,22,26.
Intestino delgado
En el intestino delgado, la enfermedad celíaca, también conocida como enteropatía por gluten, es una condición común en la que el sistema inmunitario responde anormalmente al gluten, una proteína encontrada en algunos alimentos, de modo que se produce el daño de la pared del intestino delgado5,27. La enfermedad celíaca puede afectar hasta 1 de 300 individuos, pero puede ser hasta 10 veces más prevalente en pacientes con SS que en la población general. El incremento de la prevalencia de la enfermedad celíaca se ha visto en otras condiciones mediadas por el sistema inmunitario como en la tiroiditis autoinmune o diabetes tipo 1. Los síntomas de la enfermedad celíaca incluyen diarrea, estreñimiento, fatiga, pérdida de peso, flatulencia y malestar abdominal, condiciones asociadas con deficiencia de nutrientes y vitaminas que conllevan frecuentemente a osteoporosis y anemia.
El diagnóstico de la enfermedad celíaca es difícil si se basa en síntomas solamente, por lo cual deben realizarse exámenes de autoanticuerpos gastrointestinales, los cuales deben ser solicitados en pacientes con síntomas gastrointestinales o deficiencias nutricionales. La EVDA puede demostrar alteraciones en las vellosidades y cambios histológicos; la eliminación del gluten típicamente permite la resolución del daño del intestino delgado28,29. Dentro de otros compromisos intestinales, aunque es muy raro, puede presentarse infarto intestinal o isquemia intestinal por vasculitis cuando el SS está asociado a crioglobulinemia30.
Colon
El compromiso del colon como manifestación extraglandular de SS no ha sido bien caracterizado y todavía no es claro en qué grado puede llegar a afectarlo. El estreñimiento y la diarrea pueden ocurrir frecuentemente, y se ha descrito su asociación con síndrome de intestino irritable (SII)27, enfermedad inflamatoria intestinal (EII)31, enfermedad diverticular, adenomas colónicos, tumores malignos26 y, más recientemente, con disbiosis intestinal32. Una gran proporción de pacientes con SS presenta los síntomas típicos del SII o de la dismotilidad, así como también se ha descrito que el complejo seco puede representar una manifestación extraintestinal del SII33, y hay dudas acerca de la asociación verdadera del SII con SS. En cuanto a enfermedad diverticular, esta no tiene relación directa con SS; sin embargo, no se puede excluir la posibilidad de que se produzcan con mayor frecuencia en SS, favorecida por el estreñimiento, debido, por un lado, a la propia enfermedad y, por otro lado, al uso de medicamentos antiinflamatorios27.
También se sabe que la disbiosis intestinal es un problema prevalente en SS, que llega a superar el 20% de los casos34, la disbiosis intestinal se ve favorecida por el compromiso de la integridad del epitelio y su función de barrera en SS, lo que posiblemente dé lugar a una interacción perturbada del sistema inmunitario microbiota-huésped, y puede tanto reducir como amplificar la enfermedad inflamatoria local y sistémica35. Incluso se han reportado bajos niveles de bacterias de los géneros Alistipes y Bifidobacterium, y una correlación moderada de los niveles séricos de calprotectina fecal con actividad de la enfermedad; si es > 150 μg/g, puede indicar una enfermedad orgánica concomitante (por ejemplo, una neoplasia o una EII) y amerita de estudios adicionales34.
Páncreas
La pancreatitis puede ser una consecuencia de la misma inflamación autoinmune que afecta la glándula salival y lacrimal. La pancreatitis autoinmune es una complicación rara en SS, se caracteriza por inflamación difusa del páncreas y del estrechamiento del ducto pancreático principal, lo que conlleva dolor abdominal, náuseas y emesis; esta condición se ha descrito como pancreatitis primaria y puede estar asociada a otra condición autoinmune. La elevación de los niveles de IgG4 ayuda a confirmar el diagnóstico de pancreatitis autoinmune tipo 1. La correlación entre niveles elevados de IgG4 y pacientes con SS puede actualmente ser una enfermedad sistémica asociada a sialoadenitis esclerosante, CEP, fibrosis retroperitoneal o aortitis no infecciosa. Un diagnóstico temprano puede prevenir el desarrollo de pancreatitis crónica e insuficiencia pancreática36,37.
Sistema hepatobiliar
Hasta el 7% de los pacientes con SS pueden tener algún grado de compromiso hepático, usualmente con elevación de enzimas hepáticas que cursan de manera subclínica y asintomática. En estos pacientes, las biopsias hepáticas demuestran características de CBP, HAI o CEP5,38.
La CBP es una enfermedad autoinmune del hígado, asociada con SS, en la cual los pequeños conductos biliares son atacados y destruidos por el sistema inmunitario. La inflamación continua y el daño de estos conductos biliares permiten la cicatrización y producción de cirrosis e insuficiencia hepática. En SS, la elevación de las pruebas de función hepática, la detección de los AMA, sugieren el diagnóstico de CBP; sin embargo, la biopsia generalmente es necesaria para confirmar este compromiso39.
Aproximadamente el 40%-50% de los pacientes con CBP van a tener síntomas secos que mimetizan el SS, incluidas las xeroftalmia y la xerostomía7,40. El prurito en la piel es uno de los síntomas más comunes en la CBP, que en casos leves puede ser manejado con emolientes, mientras que en casos graves, la adición de resinas no absorbentes como la colestiramina o colestipol pueden brindar un control del prurito. Estas resinas ligan muchas sustancias en el tracto gastrointestinal e inhiben la reabsorción de sales biliares5.
Otros síntomas de la complicación de la CBP incluyen la hiperpigmentación, artritis, enfermedad metabólica del hueso (osteoporosis y osteomalacia) y deficiencia de vitaminas liposolubles; en estados tardíos, cuando ya hay cirrosis se incluyen ictericia, edema, hipertensión portal y ascitis5.
En cuanto al tratamiento, el único medicamento que puede modificar la historia natural y progresión de la CBP es la administración de UDCA41. Cuando ya hay falla terapéutica y progresión de la enfermedad, se considera la realización de trasplante hepático39,42.
La HAI está caracterizada por inflamación crónica que eventualmente puede llegar a cirrosis, la mayoría de pacientes con HAI la presentan con síntomas inespecíficos como fatiga, letargia, hiporexia y artralgias; raramente la HAI presenta insuficiencia hepática aguda. La HAI se sospecha en pacientes con SS que desarrollan un aumento progresivo de enzimas hepáticas sin otra explicación, como debido a medicamentos. Esta entidad es diagnosticada con la combinación de autoanticuerpos y características específicas de la biopsia hepática. No todos los pacientes con HAI necesitan tratamiento, la decisión de iniciarlo depende de la gravedad de la enfermedad e incluye glucocorticoides e inmunomoduladores como la azatioprina40.
La CEP es una hepatopatía colestásica crónica que se caracteriza por la presencia de alteraciones estructurales de los conductos biliares tanto intrahepáticos como extrahepáticos, en los que prevalece la fibrosis obliterante; es una patología claramente autoinmune que se asocia frecuentemente a autoanticuerpos positivos dentro de los cuales se reconocen los ANA y p-ANCA. Existen pocos casos reportados en la literatura que han descrito la presencia de SS y CEP, en los que se han encontrado características comunes de la presentación del cuadro, como dolor abdominal, ictericia y diarrea7, y se asocia a la presencia de otras patologías como pancreatitis crónica y procesos autoinmunes como la fibrosis retroperitoneal, lo cual puede favorecer la sospecha clínica y, a su vez, un diagnóstico temprano de CEP en pacientes con SS; también es importante indagar acerca de otras enfermedades relacionadas con IgG4 en pacientes con la coexistencia de estas enfermedades, como la pancreatitis autoinmune o fibrosis retroperitoneal6.
La infección por el virus de la hepatitis C no es más prevalente en SS con respecto a la población general, aunque esta infección puede producir un síndrome similar a Sjögren, dado por síndrome seco. La elevación de las enzimas hepáticas en la infección por hepatitis C produce xerostomía y xeroftalmia, artralgias y sialoadenitis linfocítica que mimetiza la clásica presentación de Sjögren; sin embargo, aunque el virus de la hepatitis C puede producir ANA y factor reumatoide positivo, no se tornan positivos los anticuerpos anti-Ro ni anti-La, lo cual hace que se diferencien estas dos entidades patológicas43,44.
Discusión
Algunas características del cuadro presentado llaman la atención y merecen ser analizadas a la luz del conocimiento actual.
La paciente cursó de modo asintomático y se presentó con perfil colestásico, y registró valores de fosfatasa alcalina más de tres veces por encima de lo normal sin eosinofilia, por lo que se procedió a realizar una CRM que confirmó el diagnóstico de CEP, mientras que la IgG4 y los AMA resultaron negativos. En CEP, alrededor del 50% de los pacientes son asintomáticos en la presentación inicial con un historial de química hepática anormal45. La visualización del árbol biliar es la piedra angular en el diagnóstico y la CPRE es el método de preferencia para establecerlo; se han descrito la estenosis en banda y las formaciones arrosariadas como altamente específicas de CEP. Sin embargo, la CRM es la primera prueba de elección, al no ser invasiva y tener precisión diagnóstica comparable a la CPRE45, con una sensibilidad del 86% y especificidad del 94% para el diagnóstico de CEP41,46. La evaluación de los AMA permitirá diferenciar de CBP, ya que en CEP estarán ausentes47. La medición de IgG4 en suero (un marcador característico de la pancreatitis autoinmune que se observa en niveles más bajos en varias otras enfermedades) permitirá discernir la colangitis asociada a IgG4 de la CEP48, y se ha descrito que hasta el 9% de los pacientes pueden presentar valores por encima del límite superior de la normalidad49.
Aunque existe una fuerte asociación de la CBP con el SS, se han descrito muy pocos casos de SS asociados a CEP, y esta asociación es considerada esporádica. Cuando se han visto asociadas suelen presentarse con pancreatitis crónica. La hiposecreción pancreática es un hallazgo característico en SS y puede llegar a ser anticipatorio en CBP; sin embargo, no está claro que pueda existir en los pacientes portadores de SS asociado a CEP50. No obstante, cabe mencionar que en la CEP puede haber un grupo diverso de enfermedades y que pueden intervenir distintas etiologías, lo que conduce a un cuadro clínico común51. En SS al menos un tercio de los pacientes desarrolla manifestaciones extraglandulares de la enfermedad (más allá de las glándulas salivales y lagrimales), lo que da lugar a un espectro clínico amplio debido a las numerosas manifestaciones sistémicas posibles, hallazgos serológicos y posibles complicaciones14. En el paciente de este caso se confirmó SS, ANA positivo con patrón moteado, con anticuerpos anti-Ro y anti-La positivos y biopsia de glándula salival compatibles, sin hallazgos para alguna otra enfermedad del tejido conectivo; desde el punto de vista hepatobiliar también cursó de modo asintomático y al momento del diagnóstico de CEP no había clínica que sugiriera pancreatitis crónica.
En la patogenia de la CEP las hipótesis sobre el desencadenamiento inicial incluyen infección u otro estímulo inmunológico en individuos con predisposición genética subyacente, lesión inmunológica, lesiones vasculares, entre otras52. El proceso fisiopatológico de la inflamación y los procesos fibróticos puede estar en curso durante años antes de la aparición clínica6,50,52. En SS se ha propuesto que en la patogénesis de enfermedad sistémica la mayoría de las manifestaciones extraglandulares se consideran expresión de epitelitis autoinmune, ya que el componente epitelial es el objetivo principal de la respuesta autoinmune3,53. En el caso presentado, la asociación de SS y CEP sugiere que se presentó un mecanismo autoinmune subyacente, al tratarse de una paciente con manifestaciones sicca generalizado con xerostomía y xeroftalmia (ojo seco) con trastorno concomitante como la CEP.
A pesar de haber presentado hallazgos de perfil hepático colestásico, anormalidades colangiográficas y niveles altos de ANA circulantes, cursó sin prurito, sin niveles circulantes elevados de IgG o anticuerpos antimúsculo liso, tampoco se documentó colitis ulcerativa (CU) en toda la evolución de la enfermedad, por lo que no fue conclusivo para sobreposición. Su tratamiento inicial incluyó UDCA y no hubo requerimiento de inmunosupresión. En el estudio de Floreani y colaboradores54 se compararon las características clínicas de 7 pacientes con sobreposición HAI-CEP con las de 34 pacientes con CEP clásica y se encontró que los pacientes con sobreposición de CEP-HAI tenían edades más tempranas en su presentación (media de edad de 21 frente a 32 años), aminotransferasas significativamente más altas, características clínicas iniciales de HAI en el momento de su presentación y diagnóstico posterior de CEP cuando la inmunosupresión no logró la remisión clínica, además de prevalencia de autoanticuerpos y niveles de inmunoglobulina sérica. La CEP también es más frecuente en pacientes de mediana edad con EII, y los hombres representan el 60%-70% de los pacientes con CU y CEP41.
A partir de tres metaanálisis se sabe que el UDCA en dosis de hasta 15 mg/kg/día se ha asociado a una mejoría de bioquímica hepática y estabilización de la inflamación hepática, pero sin influir en desenlaces clínicos a largo plazo, que incluyen supervivencia y retraso en la necesidad de trasplante hepático8,9,55. El tratamiento con dosis altas (28 a 30 mg/kg al día) se asocia al incremento del riesgo de desarrollo de várices, muerte y trasplante hepático en los siguientes 5 años41,42. Una directriz de 2010 emitida por la Asociación Estadounidense para el Estudio de Enfermedades Hepáticas (AASLD) desaconseja el uso del UDCA para el tratamiento específico de la CEP56, mientras que, en 2015, El Colegio Estadounidense de Gastroenterología (ACG) no hizo ninguna recomendación sobre el uso de UDCA, aparte de señalar que no debe utilizarse en dosis superiores a 28 mg/kg/día41. En este sentido, en CEP, el uso de UDCA está dirigido a controlar la colestasis, y aunque la dosis estándar de UDCA no se ha asociado a resultados adversos, no puede recomendarse de forma rutinaria hasta que se disponga de más datos sobre eficacia y seguridad.
Ante la documentación de estenosis biliar en la CPRE, se puede llegar requerir de dilatación endoscópica con o sin colocación de endoprótesis41,45. Los corticosteroides y otros agentes inmunosupresores se recomiendan en pacientes con síndromes de sobreposición41. Una revisión Cochrane con 13 tratamientos evaluados en 22 ensayos aleatorios con 2211 participantes no demostró eficacia del tratamiento médico en CEP57. El trasplante hepático es el único tratamiento que ha demostrado prolongar la supervivencia42.
Conclusiones
Cualquier parte del sistema gastrointestinal puede ser afectado en SS, debe considerarse el rol de los medicamentos, infecciones, dieta, nutrición y lesiones tumorales.
La asociación de CEP y SS es infrecuente, se define de causa autoinmune y su tratamiento depende del componente de CEP.
Como muestra este caso, la CEP puede cursar de modo silente en SS, y procedimientos sencillos como la medición del perfil hepático y ulterior CRM pueden llevar a identificarla.
Agradecimientos
Ninguna declarada por los autores.
REFERENCIAS
1. Ramos-Casals M, Brito-Zerón P, Sisó-Almirall A, Bosch X. Primary Sjögren syndrome. BMJ. 2012;345(7872):22700787. https://doi.org/10.1136/bmj.e3821 [ Links ]
2. Vivino FB. Sjogren’s syndrome: Clinical aspects. Clin Immunol. 2017;182:48-54. https://doi.org/10.1016/j.clim.2017.04.005 [ Links ]
3. Tincani A, Andreoli L, Cavazzana I, Doria A, Favero M, Fenini MG, et al. Novel aspects of Sjögren’s syndrome in 2012. BMC Med. 2013;11:93. https://doi.org/10.1186/1741-7015-11-93 [ Links ]
4. Negrini S, Emmi G, Greco M, Borro M, Sardanelli F, Murdaca G, et al. Sjögren’s syndrome: a systemic autoimmune disease. Clin Exp Med. 2022;22(1):9-25. https://doi.org/10.1007/s10238-021-00728-6 [ Links ]
5. Ebert EC. Gastrointestinal and hepatic manifestations of Sjogren syndrome. J Clin Gastroenterol. 2012;46(1):25-30. https://doi.org/10.1097/MCG.0b013e3182329d9c [ Links ]
6. Zeron PB, Retamozo S, Bové A, Kostov BA, Sisó A, Ramos-Casals M. Diagnosis of liver involvement in primary sjögren syndrome. J Clin Transl Hepatol. 2013;1(2):94-102. https://doi.org/10.14218/JCTH.2013.00011 [ Links ]
7. Ramos-Casals M, Sánchez-Tapias JM, Parés A, Forns X, Brito-Zerón P, Nardi N, et al. Characterization and differentiation of autoimmune versus viral liver involvement in patients with Sjögren’s syndrome. J Rheumatol. 2006;33(8):1593-9. [ Links ]
8. Shi J, Li Z, Zeng X, Lin Y, Xie WF. Ursodeoxycholic acid in primary sclerosing cholangitis: Meta-analysis of randomized controlled trials. Hepatol Res. 2009;39(9):865-73. https://doi.org/10.1111/j.1872-034X.2009.00527.x [ Links ]
9. Poropat G, Giljaca V, Stimac D, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database Syst Rev. 2011;2011(1):CD003626. https://doi.org/10.1002/14651858.CD003626.pub2 [ Links ]
10. Reksten TR, Jonsson MV. Sjögren’s Syndrome: An update on epidemiology and current insights on pathophysiology. Oral Maxillofac Surg Clin North Am. 2014;26(1):1-12. https://doi.org/10.1016/j.coms.2013.09.002 [ Links ]
11. Dawson LJ, Allison HE, Stanbury J, Fitzgerald D, Smith PM. Putative anti-muscarinic antibodies cannot be detected in patients with primary Sjögren’s syndrome using conventional immunological approaches. Rheumatology. 2004;43(12):1488-95. https://doi.org/10.1093/rheumatology/keh389 [ Links ]
12. Papas AS, Sherrer YS, Charney M, Golden HE, Medsger TA, Walsh BT, et al. Successful treatment of dry mouth and dry eye symptoms in Sjögren’s syndrome patients with oral pilocarpine: A randomized, placebo-controlled, dose-adjustment study. J Clin Rheumatol. 2004;10(4):169-77. https://doi.org/10.1097/01.rhu.0000135553.08057.21 [ Links ]
13. Mandl T, Ekberg O, Wollmer P, Manthorpe R, Jacobsson LTH. Dysphagia and dysmotility of the pharynx and oesophagus in patients with primary Sjögren’s syndrome. Scand J Rheumatol. 2007;36(5):394-401. https://doi.org/10.1080/03009740701607638 [ Links ]
14. Leone MC, Alunno A, Cafaro G, Valentini V, Marcucci E, Bartoloni E, et al. The clinical spectrum of primary sjögren’s syndrome: Beyond exocrine glands. Reumatismo. 2017;69(3):93-100. https://doi.org/10.4081/reumatismo.2017.1032 [ Links ]
15. Waterman SA, Gordon TP, Rischmueller M. Inhibitory effects of muscarinic receptor autoantibodies on parasympathetic neurotransmission in Sjogren’s syndrome. Arthritis Rheum. 2000;43(7):1647-54. https://doi.org/10.1002/1529-0131(200007)43:7<1647::AID-ANR31>3.0.CO;2-P [ Links ]
16. Kjellén G, Fransson SG, Lindström F, Sökjer H, Tibbling L. Esophageal function, radiography, and dysphagia in Sjögren’s syndrome. Dig Dis Sci. 1986;31(3):225-9. https://doi.org/10.1007/BF01318111 [ Links ]
17. Grande L, Lacima G, Ros E, Font J, Pera C. Esophageal Motor Function in Primary Sjögren’s Syndrome. Am J Gastroenterol. 1993;88(3):378-81. [ Links ]
18. Türk T, Pirildar T, Tunç E, Bor S, Doğanavşargil E. Manometric assessment of esophageal motility in patients with primary Sjögren›s syndrome. Rheumatol Int. 2005;25(4):246-9. https://doi.org/10.1007/s00296-003-0426-9 [ Links ]
19. Rosztóczy A, Kovács L, Wittmann T, Lonovics J, Pokorny G. Manometric assessment of impaired esophageal motor function in primary Sjögren›s syndrome. Clin Exp Rheumatol. 2001;19(2):147-52. [ Links ]
20. Parra-Izquierdo V, Bernal Macías S, Avila F, Costa V, Leguízamo A, Hani A. Serie de casos de pacientes con enfermedad de tejido conectivo no esclerodermia con alteraciones motoras esofágicas y anorrectales por manometría de alta resolución en el Hospital Universitario San Ignacio durante el 2016. Bucaramanga, Colombia: XVI Congreso Colombiano de Reumatología; 2017. [ Links ]
21. Belafsky PC, Postma GN. The laryngeal and esophageal manifestations of Sjögren’s syndrome. Curr Rheumatol Rep. 2003;5(4):297-303. https://doi.org/10.1007/s11926-003-0008-6 [ Links ]
22. El Miedany YM, Baddour M, Ahmed I, Fahmy H. Sjogren’s syndrome: Concomitant H. Pylori infection and possible correlation with clinical parameters. Jt Bone Spine. 2005;72(2):135-41. https://doi.org/10.1016/j.jbspin.2004.04.005 [ Links ]
23. Buchanan WW, Cox AG, Harden RM, Glen AI, Anderson JR, Gray KG. Gastric studies in sjögren’s syndrome. Gut. 1966;7(4):351-4. https://doi.org/10.1136/gut.7.4.351 [ Links ]
24. Kilpi A, Bergroth V, Konttinen YT, Maury CPJ, Reitamo S, Wegelius O. Lymphocyte infiltrations of the gastric mucosa in sjöugren’s syndrome. An Immunoperoxidase Study Using Monoclonal Antibodies in the Avidin‐Biotin‐Peroxidase Method. Arthritis Rheum. 1983;26(10):1196-200. https://doi.org/10.1002/art.1780261004 [ Links ]
25. Maury CPJ, Törnroth T, Teppo A‐M. Atrophic gastritis in sjögren’s syndrome. Morphologic, biochemical, and immunologic findings. Arthritis Rheum. 1985;28(4):388-94. https://doi.org/10.1002/art.1780280406 [ Links ]
26. Brito-Zerón P, Kostov B, Fraile G, Caravia-Durán D, Maure B, Rascón FJ, et al. Characterization and risk estimate of cancer in patients with primary Sjögren syndrome. J Hematol Oncol. 2017;10(1):17-8. https://doi.org/10.1186/s13045-017-0464-5 [ Links ]
27. Kim-Lee C, Suresh L, Ambrus JL. Gastrointestinal disease in Sjogren’s syndrome: related to food hypersensitivities. Springerplus. 2015;4(1):1-5. https://doi.org/10.1186/s40064-015-1557-7 [ Links ]
28. Iltanen S, Collin P, Korpela M, Holm K, Partanen J, Polvi A, et al. Celiac Disease and Markers of Celiac Disease Latency in Patients With Primary Sjögren’s Syndrome. Am J Gastroenterol. 1999;94(4):1042-6. https://doi.org/10.1111/j.1572-0241.1999.01011.x [ Links ]
29. Szodoray P, Barta Z, Lakos G, Szakáll S, Zeher M. Coeliac disease in Sjögren’s syndrome - A study of 111 Hungarian patients. Rheumatol Int. 2004;24(5):278-82. https://doi.org/10.1007/s00296-003-0360-x [ Links ]
30. Doyle MK. Vasculitis associated with connective tissue disorders. Curr Rheumatol Rep. 2006;8(4):312-6. https://doi.org/10.1007/s11926-006-0015-5 [ Links ]
31. Palm Ø, Moum B, Gran JT. Estimation of Sjögren’s syndrome among IBD patients: A six year post-diagnostic prevalence study. Scand J Rheumatol. 2002;31(3):140-5. https://doi.org/10.1080/rhe.31.3.140.145 [ Links ]
32. De Paiva CS, Jones DB, Stern ME, Bian F, Moore QL, Corbiere S, et al. Altered Mucosal Microbiome Diversity and Disease Severity in Sjögren Syndrome. Sci Rep. 2016;6: 23561. https://doi.org/10.1038/srep23561 [ Links ]
33. Barton A, Pal B, Whorwell PJ, Marshall D. Increased prevalence of sicca complex and fibromyalgia in patients with irritable bowel syndrome. Am J Gastroenterol. 1999;94(7):1898-901. https://doi.org/10.1111/j.1572-0241.1999.01146.x [ Links ]
34. Mandl T, Marsal J, Olsson P, Ohlsson B, Andréasson K. Severe intestinal dysbiosis is prevalent in primary Sjögren’s syndrome and is associated with systemic disease activity. Arthritis Res Ther. 2017;19(1):237. https://doi.org/10.1186/s13075-017-1446-2 [ Links ]
35. Forbes JD, Van Domselaar G, Bernstein CN. The gut microbiota in immune-mediated inflammatory diseases. Front Microbiol. 2016;7:1081. https://doi.org/10.3389/fmicb.2016.01081 [ Links ]
36. Hayakawa T, Naruse S, Kitagawa M, Kondo T. Clinical aspects of autoimmune pancreatitis in Sjogren’s syndrome. J Pancreas. 2001;2(3):88-92. [ Links ]
37. Afzelius P, Fallentin EM, Larsen S, Møller S, Schiødt M. Pancreatic function and morphology in Sjögren›s syndrome. Scand J Gastroenterol. 2010;45(6):752-8. https://doi.org/10.3109/00365521003642542 [ Links ]
38. Skopouli FN, Barbatis C, Moutsopoulos HM. Liver involvement in primary sjögren›s syndrome. Rheumatology. 1994;33(8):745-8. https://doi.org/10.1093/rheumatology/33.8.745 [ Links ]
39. Tsianos EV, Hoofnagle JH, Fox PC, Alspaugh M, Jones EA, Schafer DF, et al. Sjögren›s syndrome in patients with primary biliary cirrhosis. Hepatology. 1990;11(5):730-4. https://doi.org/10.1002/hep.1840110504 [ Links ]
40. Teufel A, Weinmann A, Kahaly GJ, Centner C, Piendl A, Wörns M, et al. Concurrent autoimmune diseases in patients with autoimmune hepatitis. J Clin Gastroenterol. 2010;44(3):208-13. https://doi.org/10.1097/MCG.0b013e3181c74e0d [ Links ]
41. Lindor KD, Kowdley KV, Harrison ME. ACG clinical guideline: Primary sclerosing cholangitis. Am J Gastroenterol. 2015;110(5):646-59. https://doi.org/10.1038/ajg.2015.112 [ Links ]
42. Martin P, Dimartini A, Feng S, Brown R, Fallon M. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Hepatology. 2014;59(3):1144-65. https://doi.org/10.1002/hep.26972 [ Links ]
43. Haddad J, Trinchet JC, Pateron D, Mal F, Beaugrand M, Munz-Gotheil C, et al. Lymphocytic sialadenitis of Sjögren›s syndrome associated with chronic hepatitis C virus liver disease. Lancet. 1992;339(8789):321-3. https://doi.org/10.1016/0140-6736(92)91645-O [ Links ]
44. Ramos-Casals M, García-Carrasco M, Cervera R, Font J. Sjogren›s syndrome and hepatitis C virus. Clin Rheumatol. 1999;18(2):93-100. https://doi.org/10.1007/s100670050064 [ Links ]
45. Aabakken L, Karlsen TH, Albert J, Arvanitakis M, Chazouilleres O, Dumonceau JM, et al. Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline. Endoscopy. 2017;49(6):588-608. https://doi.org/10.1055/s-0043-107029 [ Links ]
46. Dave M, Elmunzer BJ, Dwamena BA, Higgins PDR. Primary sclerosing cholangitis: Meta-analysis of diagnostic performance of MR cholangiopancreatography. Radiology. 2010;256(2):387-96. https://doi.org/10.1148/radiol.10091953 [ Links ]
47. Angulo P, Peter JB, Gershwin ME, DeSotel CK, Shoenfeld Y, Ahmed AEE, et al. Serum autoantibodies in patients with primary sclerosing cholangitis. J Hepatol. 2000;32(2):182-7. https://doi.org/10.1016/S0168-8278(00)80061-6 [ Links ]
48. Hirano K, Kawabe T, Yamamoto N, Nakai Y, Sasahira N, Tsujino T, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta. 2006;367(1-2):181-4. https://doi.org/10.1016/j.cca.2005.11.031 [ Links ]
49. Mendes FD, Jorgensen R, Keach J, Katzmann JA, Smyrk T, Donlinger J, et al. Elevated serum IgG4 concentration in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2006;101(9):2070-5. https://doi.org/10.1111/j.1572-0241.2006.00772.x [ Links ]
50. Hernández-Molina G, Michel-Peregrina ML. Afección pancreática en el síndrome de sjögren. Reumatol Clin. 2011;7(2):130-4. https://doi.org/10.1016/j.reuma.2010.07.005 [ Links ]
51. Guerrero P, Martín M, Conde JM, Castro M, Rodríguez MC, Castilla L, et al. Colangitis esclerosante primaria segmentaria asociada a síndrome de Sjögren. Rev Esp Enferm Dig. 1991;79(5):363-6. [ Links ]
52. Karlsen TH, Schrumpf E, Boberg KM. Primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2010;24(5):655-66. https://doi.org/10.1016/j.bpg.2010.07.005 [ Links ]
53. Scofield RH. Vasculitis in Sjögren’s syndrome. Curr Rheumatol Rep. 2011;13(6):482-8. https://doi.org/10.1007/s11926-011-0207-5 [ Links ]
54. Floreani A, Rizzotto ER, Ferrara F, Carderi I, Caroli D, Blasone L, et al. Clinical course and outcome of autoimmune hepatitis/primary sclerosing cholangitis overlap syndrome. Am J Gastroenterol. 2005;100(7):1516-22. https://doi.org/10.1111/j.1572-0241.2005.41841.x [ Links ]
55. Triantos CK, Koukias NM, Nikolopoulou VN, Burroughs AK. Meta-analysis: Ursodeoxycholic acid for primary sclerosing cholangitis. Aliment Pharmacol Ther. 2011;34(8):901-10. https://doi.org/10.1111/j.1365-2036.2011.04822.x [ Links ]
56. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51(2):660-78. https://doi.org/10.1002/hep.23294 [ Links ]
57. Saffioti F, Gurusamy KS, Hawkins N, Toon CD, Tsochatzis E, Davidson BR, et al. Pharmacological interventions for primary sclerosing cholangitis. Cochrane Database Syst Rev. 2017;2017(4):3-5. https://doi.org/10.1002/14651858.CD011343.pub2 [ Links ]
Citación: Parra-Izquierdo V, Frías-Ordóñez JS, Ovalle- Hernández AF, Costa-Barney VA, Flórez-Sarmiento C, Hani A. Compromiso gastrointestinal y hepático en síndrome de Sjögren primario: reporte de caso y revisión de la literatura. Revista. colomb. Gastroenterol. 2023;38(3):338-347. https://doi.org/10.22516/25007440.918
Aprobación ética y consentimiento de participación Se obtuvo el consentimiento informado de los pacientes en cada institución participante
Disponibilidad de datos y material Los datos y el material disponibles para la publicación están en el manuscrito y no se omite ninguna información
Recibido: 31 de Mayo de 2022; Aprobado: 13 de Julio de 2022
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
