











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink
Introduction
Neurological disorders associated with liver diseases have attracted increasing interest. Acquired hepatolenticular degeneration (ADH) was described in 1919 by Woerkem1 and was later called acquired non-Wilsonian hepatolenticular degeneration by Víctor et al.2 in 1965. It is a neurodegenerative disorder that affects the basal nuclei and has mechanisms responsible for neuronal dysfunction and death other than Wilson’s disease, which mainly generates extrapyramidal symptoms such as tremor and rigidity associated with cognitive impairment in patients with cirrhosis in the absence of Kayser-Fleischer rings characteristic of Wilson’s disease3,4. Its origin depends on deposits formed by toxic substances not eliminated by the hepatobiliary system, which allows these agents, especially heavy metals such as manganese, to be released into the circulation and play an essential role in the pathogenesis of ADH5-8.
We present the case of a man whose diagnosis of ADH was challenging, given that he had an established history of liver disease associated with alcohol consumption; however, the ammonium levels and the upper gastrointestinal endoscopy without stigmata of variceal bleeding led to thinking about different causes of encephalopathy, and magnetic resonance imaging (MRI) helped reach a definitive diagnostic approach.
Case presentation
A 55-year-old man from the United States, where esophageal variceal ligation was performed in November 2021 related to the diagnosis at the time of liver cirrhosis secondary to alcohol intake since the age of 35, drank a bottle a day on average for ten years. He had no follow-up or outpatient treatment, nor did he suffer from other comorbidities or family history. He had a complete vaccination schedule against COVID-19.
He consulted the emergency service in July 2022 due to a three-day picture of watery stools without mucus, blood, melena, or temperature rises, two episodes of small hematemesis, temporal and spatial disorientation, and changes in behavior involving aggressiveness. In the first evaluation, he was alert, dyspraxic, disoriented in time and personal identity, uncooperative with the interview, and with no alteration in vital signs. Given the history of liver disease, it was classified as West Haven 2 hepatic encephalopathy (HE) during the first hours of care. He became agitated and aggressive, with tachycardia and tachypnea. After 24 hours, he had significant neurological deterioration (HE West Haven 4), requiring orotracheal intubation to protect the airway.
His initial studies showed normocytic anemia, mild electrolyte disorder due to hypernatremia and hyperchloremia, altered liver function with cirrhosis classified as Child-Pugh B, MELD of 14, preserved renal function, increased inflammatory markers, metabolic acidemia with elevated anion gap (17.3 mEq). /L), uncompensated (arterial gases: pH 7.3, partial pressure of carbon dioxide (PCO2) of 21.8 mm Hg, partial pressure of oxygen (PO2) of 97.8 mm Hg, oxygen saturation (SatO2) of 98.1%, fraction inspired oxygen (FIO2) of 21%, bicarbonate (HCO3) of 16.6 mmol/L, Be of -5.3 mmol/L and lactic acid of 2.23 mmol/L) (Table 1).
Table 1 Paraclinical tests performed during the patient’s hospitalization
| Laboratory | Patient value | Reference value | Laboratory | Patient value | Reference value |
|---|---|---|---|---|---|
| Leukocytes | 8.04 | 4.8-11/mm3 | AST | 47 | 8-33 U/L |
| Neutrophils | 4.58 | 2.2-7.7/mm3 | ALT | 18 | 4-36 U/L |
| Lymphocytes | 2.66 | 1.3-2.9/mm3 | GGT | 35 | 5-40 U/L |
| Hemoglobin | 10.4 | 13-16 g/dL | Alkaline phosphatase | 77 | 44-147 UI/L |
| Hematocrit | 29.7 | 36 %-48 % | TB | 1.97 | 0.1-1 mg/dL |
| MCV | 93.4 | 80-100 fL | DB | 1.33 | <0.3 mg/dL |
| Platelets | 151 | 150-450/mm3 | IB | 0.64 | 0.1-0.5 mg/dL |
| Sodium | 147 | 135-145 mEq/L | Albumin | 3.3 | 3.8-5 g/dL |
| Potassium | 3.9 | 3.9-5.5 mEq/L | PT | 15.8 | 9.5-12.5 s |
| Chlorine | 117 | 98-107 mEq/L | TPT | 31.8 | 25-37 s |
| Magnesium | 1.89 | 1.6-2.4 mEq/L | Creatinine | 0.59 | 0.7-1.17 mg/dL |
| Phosphorus | 2.5 | 2.5-4.5 mg/dL | Ureic nitrogen | 19.1 | 20 mg/dL |
| Ionic calcium | 1.18 | 4.8-5.6 mg/dL | CRP | 12.5 | <1 mg/dL |
| TSH | 0.67 | 0.37-4.7 uU/mL | Procalcitonin | <0.05 | <0.5 ng/mL |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; CRP: C-reactive protein; DB: direct bilirubin; GGT: gamma-glutamyl transferase; IB: indirect bilirubin; MCV: mean corpuscular volume; PT: prothrombin time; PTT: thromboplastin time; TB: total bilirubin; TSH: thyroid stimulating hormone. Prepared by the authors.
Given the clinical and laboratory signs of a systemic inflammatory response and the probability of a systemic response, antibiotic management was indicated with ceftriaxone 1 g every 12 hours and intravenous vasoactive support with terlipressin 2 mg every 4 hours. Complementary paraclinical tests showed normal ammonium (48 µmol/L), normal liver infectious tests, normal ferrokinetic profile (ferritin 86.2 ng/mL, transferrin saturation percentage of 25%, normal vitamin B12 and folic acid, and negative hepatitis A IgM antibodies, hepatitis B surface antigen (AgSVHB), anti-AgSVHB antibodies, total hepatitis C antibodies, human immunodeficiency virus (HIV), rapid plasma reaginin (RPR), blood cultures, urine cultures, and PCR for SARS-CoV-2.
An upper GI endoscopy was performed, noting varicose cords with varicose veins smaller than 5 mm, without stigmata of recent bleeding (Paquet III) or gastric varices, which, added to the heterogeneous hepatic echogenicity documented in the abdominal ultrasound, led to the consideration of cirrhosis of alcoholic origin (risk consumption of 88 g of alcohol, 8.7 units of standard drink for ten years until November 2021), but there was no evidence of hypertensive portal bleeding. Therefore, it was necessary to rule out nonhepatic causes of encephalopathy.
A lumbar puncture showed no abnormalities in the cytochemistry, a microbiological study for molecular infectious panel turned out negative for neuroinfection, and cytology and an immunophenotype did not exhibit suspicion of tumor involvement. Finally, a nuclear MRI was performed to observe hyperintensities in basal ganglia, bilateral caudate predominance in T1 sequences, and fluid-attenuated inversion recovery (FLAIR) suggestive of hepatolenticular degeneration (Figure 1).
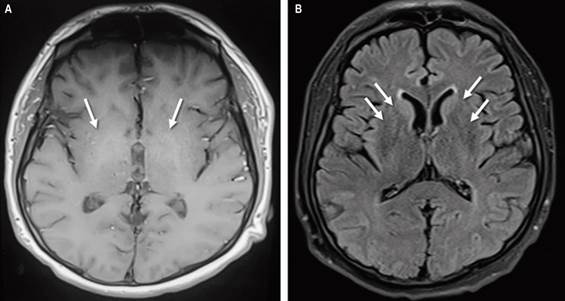
Source: Authors’ archive.
Figure 1 Brain nuclear MRI sequences T1 (A) FLAIR (B). Arrows show areas of hyperintensity in the bilateral basal ganglia.
In the search for Wilson’s disease, the ophthalmology area evaluated and ruled out the presence of Kayser-Fleischer rings. In addition, studies of serum ceruloplasmin (22 mg/dL) and urine copper (27.8 µg/24 hours) were normal.
After four days of intubation, extubation was achieved; 11 days later, he was discharged thanks to improved neurological condition, without recurrence of bleeding, stable hemoglobin, and an outpatient management plan with spironolactone, propranolol, and nutritional recommendations.
Discussion
ADH is a rare entity; in a retrospective study with a population of more than 1,000 patients with liver cirrhosis, only 0.8% had this diagnosis, and in other reports, the prevalence is even lower (less than 0.5%)5-9, while viral infectious etiology is the most common10,11. Manganese, which causes non-Wilsonian ADH, is the twelfth most crucial element and the fifth among metals12.
Several enzymatic systems depend on and interact with manganese. It is helpful for the formation of cartilage and bone, the maintenance of mitochondria, and the production of glucose13.
In in vivo and in vitro studies, both in animal models and in humans, it has been identified that SLC30A10 (Solute Carrier Family 30 Member 10), a carrier of manganese efflux from the cell, plays a vital role in the regulation of the levels of this metal. In predisposed individuals with environmental exposure due to a place of residence in industrial or mining sectors or their occupation, mutations lead to loss of function of SLC20A10 and retention of manganese in the brain, particularly in the basal ganglia and liver, which causes neurotoxicity and liver damage, respectively14. Meanwhile, pre-existing diseases and the formation of portosystemic shunts promote the release of manganese into the systemic circulation and its deposition in the brain15. A series of five cases of ADH was recently reported; four of them had cirrhosis, one had chronic hepatitis due to the hepatitis C virus, and all had portosystemic shunts16.
The clinical manifestations of the disease are very heterogeneous. Most patients have changes in behavior, Parkinsonian features, and dystonia with gait alteration17. In the case in question, the initial manifestation was a behavior change that alerted his family, then a phase of psychomotor agitation during the stay in the emergency room and subsequently a deterioration in the state of consciousness that required orotracheal intubation and invasive mechanical ventilation in the intensive care unit (ICU). These changes occur due to the increase in manganese in the systemic circulation and its underlying brain deposition, which can happen in the cortex, specifically in the basal ganglia. It entails oxidative stress and induces neurotoxicity through multiple mechanisms such as death of dopaminergic cells and alteration in γ-aminobutyric acid (GABA)-mediated transmission, both inhibitory stimuli; release of glutamate, the primary excitatory neurotransmitter, and a critical signaling molecule; choline deficiency, necessary for the synthesis of acetylcholine, and increase in acetylcholinesterase, the enzyme that degrades it13,18. Furthermore, deposits in the central nervous system lead to mitochondrial dysfunction of astrocytes at the level of benzodiazepine receptors19. This has been demonstrated in animals, in which manganese levels are observed to affect astrocytes, with a consequent decrease in high-affinity glutamate transport and stress-mediated neuronal death through the activation of the nitric oxide-cyclic guanosine monophosphate pathway18.
The diagnosis to confirm or rule out ADH requires anamnesis, physical examination findings, blood analysis, and neuroimaging such as contrast-enhanced computed tomography, and, more usually, MRI, which allows the exclusion of other diagnoses such as brain hematomas, small vessel disease, and space-occupying lesions8. In ADH, MRI reveals changes in the basal ganglia with T1 hyperintensity, especially in the globus pallidus, a significant proportion in cirrhotic patients, related to the severity of the disease20. Laboratory tests can sometimes be helpful; however, it is worth mentioning that manganese levels in blood and urine indicate recent exposure (hours to days), but there are no biomarkers of cumulative exposure to manganese nor prognostic biomarkers of its neurotoxic effects. Therefore, they are not diagnostic.
In the case of suspected Wilson’s disease, ceruloplasmin and ammonia levels can be requested to rule it out. In our patient, they were negative, which, in addition to the absence of Kayser-Fleischer rings on physical examination, allowed us to reinforce the diagnostic suspicion, which was complemented by MRI findings of manganese deposits as a causal agent of encephalopathy19. Anyway, there is a good correlation between blood manganese levels and T1 hyperintensity observed by MRI, not necessarily correlated with neurological function, so the diagnosis is ultimately based on clinical suspicion and exclusion of more frequent causes without a diagnostic test confirming another etiology16,20.
Based on recent evidence in HE West Haven 4, finding normal ammonium levels with its high pretest probability is infrequent; still, regardless of the result, the priority in acute conditions is to manage potential contributors such as gastrointestinal bleeding, electrolyte alterations, and infections21. Once clinical stability is achieved, if ammonia is normal in HE, it is essential to evaluate the etiologies causing the systemic or neuronal inflammation that resulted in the activation of microglia and encephalopathy in the diagnostic algorithm22.
For the case reported, the starting point was an already established liver cirrhosis with previous endoscopic management of esophageal varices, which required bleeding to be ruled out. Still, on this occasion, there were no stigmata of recent bleeding. Cultures to rule out systemic and central nervous system infectious causes were negative; however, we could not rule out culture-negative sepsis sensitive to ceftriaxone (community-acquired) due to metabolic acidosis, the possibility of bacterial translocation due to gastrointestinal bleeding, the clinical signs of clinical response, elevated acute inflammation reactants, and a leukocyte count of 8,040/mm3, which is higher than expected in portal hypertension and a prognostic marker for organ failure and mortality, as shown by the CANONIC study in acute-on-chronic liver failure. Therefore, antibiotics and supportive management were necessary in this patient.
Metabolic etiologies related to storage diseases were evaluated, obtaining a normal ferrokinetic profile. On the other hand, in the absence of standardized tests to measure manganese levels, normal ammonium and hyperintensities in the basal ganglia by brain MRI were vital in the differential diagnosis between Wilson’s disease and non-Wilsonian ADH, in favor of the latter, taking into account normal levels of serum ceruloplasmin and serum and urinary copper. The distinguishing characteristics of the two diseases are listed in Table 2.
Table 2 The main differences between Wilson’s disease and non-Wilsonian hepatolenticular degeneration
| Wilson’s disease | Non-Wilsonian hepatolenticular degeneration |
|---|---|
| Autosomal inheritance pattern | Acquired |
| Copper deposits in the liver and brain | Manganese deposits in the brain |
| The inability of ceruloplasmin and copper to bind | Association with liver failure, mainly of alcohol origin |
| ATP7B carrier mutation | Increased transport of manganese at the intestinal level and hepatocytes |
| Extrapyramidal signs of non-Parkinsonian predominance | Predominance of Parkinsonism and extrapyramidal syndrome |
| Multisystem condition | Histopathological findings similar to Wilson’s disease |
Adapted from: Rebolledo-García D, et al. Med Int Méx. 2015;31:478-84.
Regarding treatment, even if ADH is recognized, it is limited. The use of dopamine agonists may be considered given the symptoms similar to Parkinson’s; however, a large proportion of patients do not have a good response. The evidence for using rifaximin in managing HE in patients with ADH is poor and comes from case series. As occurred in the patient, the most relevant thing is the supportive treatment of the liver disease and nutritional measures for outpatient management.
A series of three cases of Wilsonian hepatolenticular degeneration was previously reported, and only one documented findings in the basal ganglia in the FLAIR sequence with various clinical manifestations. In contrast, in the case presented, MRI was fundamental for the final diagnosis of non-Wilsonian hepatolenticular degeneration23. This experience shows that the different ways this disease can manifest make necessary a comprehensive approach, including the clinical picture and the diagnostic studies available for each case.
Conclusion
ADH is a rare disease as a cause of encephalopathy in patients with liver disease, primarily associated with alcoholism, which produces debilitating neurological symptoms resulting from manganese deposits in the basal ganglia. For its diagnosis, it is necessary to rule out other more frequent causes and evaluate the brain image in search of the typical findings in the T1 sequence. While the etiology is defined, management should focus on supportive measures of the liver disease.

