










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista colombiana de Gastroenterología
versão impressa ISSN 0120-9957versão On-line ISSN 2500-7440
Rev. colomb. Gastroenterol. vol.38 no.4 Bogotá out./dez. 2023 Epub 26-Fev-2024
https://doi.org/10.22516/25007440.989
Reporte de casos
Manganeso asociado con degeneración hepatolenticular no wilsoniana como causa infrecuente de encefalopatía: reporte de caso
1
 http://orcid.org/0000-0003-1139-8694
http://orcid.org/0000-0003-1139-8694
1
*
http://orcid.org/0000-0002-6079-6974
1
http://orcid.org/0000-0002-2660-6671
1 Residente de Medicina Interna, Grupo de Investigación en Medicina Interna (GIMI), Universidad Libre, seccional Cali. Cali, Colombia.
2 Médico familiar, internista, geriatra, magíster en Epidemiología, Gerontología Social y Cuidados Paliativos. Docente, Universidad Libre, seccional Cali y Universidad del Valle. Cali, Colombia.
3Hepatólogo, gastroenterólogo, Departamento de Gastroenterología y Endoscopia, Clínica Farallones. Cali, Colombia.
Objetivo:
Describir la presentación clínica, el diagnóstico y el tratamiento de un paciente con encefalopatía como manifestación de degeneración hepatolenticular no wilsoniana producida por manganeso, en un centro de alta complejidad de un país latinoamericano.
Descripción del caso:
Paciente masculino de 55 años, procedente de Estados Unidos, con antecedente de enfermedad hepática asociada con consumo de alcohol, quien ingresó al servicio de urgencias por un cuadro de diarrea, hematemesis y agitación psicomotora. Durante la estancia presentó deterioro en el estado de consciencia, por lo que requirió intubación orotraqueal. En su estudio diagnóstico, las pruebas de líquido cefalorraquídeo fueron negativas para etiologías infecciosas, en los estudios endoscópicos no tenía estigmas de sangrado portal hipertensivo y el amonio y los estudios para causas metabólicas fueron normales. Sin embargo, se documentaron áreas de hiperintensidad en los ganglios de la base en la resonancia magnética cerebral, con niveles de ceruloplasmina sérica y cobre urinario normales, lo que descartó enfermedad de Wilson y definió el diagnóstico de degeneración hepatolenticular no wilsoniana por depósitos de manganeso.
Conclusión:
La degeneración hepatolenticular no wilsoniana es una causa infrecuente de encefalopatía crónica con manifestaciones clínicas de extrapiramidalismo, secundaria a disfunción de los ganglios de la base por enfermedad hepática grave. Su diagnóstico se convierte en un reto, dado que se produce por depósitos de manganeso y no existen biomarcadores que puedan establecer el nivel de exposición a este metal. La resonancia magnética cerebral juega, por tanto, un papel indispensable al reflejar esos depósitos en los ganglios de la base.
Palabras clave: Encefalopatía hepática; degeneración hepatolenticular; manganeso; diagnóstico por imagen
Aim:
To describe the clinical picture, diagnosis, and treatment of a patient with encephalopathy as a manifestation of manganese-induced non-Wilsonian hepatolenticular degeneration (NWHD) in a high-complexity care center in a Latin American country.
Case description:
A 55-year-old male patient from the United States with a history of liver disease associated with alcohol consumption was admitted to the emergency department due to diarrhea, hematemesis, and psychomotor agitation. During his stay, his state of consciousness deteriorated, requiring orotracheal intubation. In his diagnostic study, cerebrospinal fluid tests were negative for infectious etiologies; the endoscopic examinations showed no marks of portal hypertension bleeding, while ammonium and tests for metabolic causes were normal. However, areas of hyperintensity in the basal ganglia were documented on brain MRI, with normal ceruloplasmin serum and urine copper levels, which ruled out Wilson’s disease and determined the diagnosis of manganese-induced NWHD.
Conclusion:
NWHD is a rare cause of chronic encephalopathy with clinical manifestations of extrapyramidal symptoms secondary to basal ganglia dysfunction due to severe liver disease. Its diagnosis becomes a challenge, given that manganese deposits produce it, and no biomarkers can establish the level of exposure to this metal. Brain MRI is indispensable in reflecting these deposits in the basal ganglia.
Keywords: Hepatic encephalopathy; hepatolenticular degeneration; manganese; diagnostic imaging
Introducción
Los trastornos neurológicos asociados con enfermedades hepáticas han despertado un interés creciente. La degeneración hepatolenticular adquirida (DHA) fue descrita en 1919 por Woerkem1 y más tarde fue denominada como degeneración hepatolenticular adquirida no wilsoniana por Víctor y colaboradores2 en 1965. Se trata de un trastorno neurodegenerativo que afecta a los núcleos basales y tiene mecanismos responsables de la disfunción y muerte neuronal distintos a la enfermedad de Wilson, lo que genera particularmente síntomas extrapiramidales como temblor y rigidez asociados con deterioro cognitivo en pacientes con cirrosis, en ausencia de anillos de Kayser-Fleischer característicos de la enfermedad de Wilson3,4. Su origen depende de la existencia de depósitos formados por sustancias toxicas que no son eliminadas por el sistema hepatobiliar, lo que permite que estos agentes, especialmente metales pesados como el manganeso, sean liberados a la circulación y presenten un papel importante en la patogenia de la DHA5-8.
Presentamos el caso de un hombre cuyo diagnóstico de DHA fue un reto dado que tenía una historia ya establecida de enfermedad hepática asociada con consumo de alcohol; sin embargo, los niveles de amonio y la endoscopia digestiva superior sin estigmas de sangrado variceal llevaron a pensar en causas diferentes de encefalopatía, y la resonancia magnética nuclear (RMN) permitió llegar a una aproximación diagnóstica definitiva.
Presentación del caso
Hombre de 55 años, procedente de Estados Unidos, país donde se le realizó la ligadura de várices esofágicas en noviembre de 2021 relacionada con el diagnóstico en ese momento de cirrosis hepática secundaria a ingesta de alcohol desde los 35 años. En promedio bebió una botella al día por 10 años, no tenía seguimiento ni tratamiento ambulatorio, tampoco padecía otras comorbilidades, ni antecedentes familiares. Presentaba esquema de vacunación completo contra COVID-19.
Consultó al servicio de urgencias en julio de 2022 por un cuadro de tres días consistente en deposiciones acuosas, sin moco, sangre, melenas, ni alzas térmicas y dos episodios de hematemesis de escasa cantidad; además, desorientación temporal, espacial y cambios en el comportamiento dados por agresividad. En la primera evaluación se encontraba alerta, disproséxico, desorientado en tiempo y persona, poco colaborador con la entrevista y sin alteración en signos vitales. Dado el antecedente de enfermedad hepática durante las primeras horas de atención se clasificó como una encefalopatía hepática (EH) West Haven 2; inicialmente se tornó agitado, agresivo, con taquicardia y taquipnea. Luego de 24 horas tuvo un deterioro neurológico significativo (EH West Haven 4), por lo que fue necesaria la intubación orotraqueal para proteger la vía aérea.
Sus estudios iniciales mostraron anemia normocítica, trastorno hidroelectrolítico leve por hipernatremia e hipercloremia, función hepática alterada con cirrosis clasificada como Child Pugh B, MELD de 14, función renal preservada, aumento de marcadores inflamatorios, acidemia metabólica con anión GAP elevado (17,3 mEq/L), no compensada (gases arteriales: pH de 7,3, presión parcial de dióxido de carbono (PCO2) de 21,8 mm Hg, presión parcial de oxígeno (PO2) de 97,8 mm Hg, saturación de oxígeno (SatO2) de 98,1%, fracción inspirada de oxígeno (FIO2) de 21%, bicarbonato (HCO3) de 16,6 mmol/L, Be de -5,3 mmol/L y ácido láctico de 2,23 mmol/L) (Tabla 1).
Tabla 1 Paraclínicos realizados durante la hospitalización de la paciente
| Laboratorio | Valor de paciente | Valor de referencia | Laboratorio | Valor de paciente | Valor de referencia |
|---|---|---|---|---|---|
| Leucocitos | 8,04 | 4,8-11/mm3 | AST | 47 | 8-33 U/L |
| Neutrófilos | 4,58 | 2,2-7,7/mm3 | ALT | 18 | 4-36 U/L |
| Linfocitos | 2,66 | 1,3-2,9/mm3 | GGT | 35 | 5-40 U/L |
| Hemoglobina | 10,4 | 13-16 g/dL | Fosfatasa alcalina | 77 | 44-147 UI/L |
| Hematocrito | 29,7 | 36 %-48 % | BT | 1,97 | 0,1-1 mg/dL |
| VCM | 93,4 | 80-100 fL | BD | 1,33 | <0,3 mg/dL |
| Plaquetas | 151 | 150-450/mm3 | BI | 0,64 | 0,1-0,5 mg/dL |
| Sodio | 147 | 135-145 mEq/L | Albúmina | 3,3 | 3,8-5 g/dL |
| Potasio | 3,9 | 3,9-5,5 mEq/L | PT | 15,8 | 9,5-12,5 s |
| Cloro | 117 | 98-107 mEq/L | PTT | 31,8 | 25-37 s |
| Magnesio | 1,89 | 1,6-2,4 mEq/L | Creatinina | 0,59 | 0,7-1,17 mg/dL |
| Fósforo | 2,5 | 2,5-4,5 mg/dL | Nitrógeno ureico | 19,1 | 20 mg/dL |
| Calcio iónico | 1,18 | 4,8-5,6 mg/dL | PCR | 12,5 | <1 mg/dL |
| TSH | 0,67 | 0,37-4,7 uU/mL | Procalcitonina | <0,05 | <0,5 ng/mL |
ALT: alanina-aminotransferasa; AST: aspartato-aminotransferasa; BD: bilirrubina directa; BI: bilirrubina indirecta; BT: bilirrubina total; GGT: gamma-glutamil transferasa; PCR: proteína C-reactiva; PT: tiempo de protrombina; PTT: tiempo de tromboplastina; TSH: hormona estimulante de la tiroides; VCM: volumen corpuscular medio. Tabla elaborada por los autores.
Ante los signos clínicos y de laboratorio de respuesta inflamatoria sistémica y la probabilidad de respuesta sistémica se indicó manejo antibiótico con ceftriaxona de 1 g cada 12 horas y soporte vasoactivo endovenosos con terlipresina de 2 mg cada 4 horas. Los paraclínicos complementarios mostraron amonio normal (48 µmol/L), pruebas infecciosas hepáticas normales, perfil ferrocinético normal (ferritina de 86,2 ng/mL, porcentaje de saturación de transferrina de 25%), vitamina B12 y ácido fólico normales; anticuerpos IgM antihepatitis A, antígeno de superficie de hepatitis B (AgSVHB), anticuerpos anti-AgSVHB, anticuerpos totales contra hepatitis C, virus de la inmunodeficiencia humana (VIH), reaginina plasmática rápida (RPR), hemocultivos, urocultivo y PCR para SARS-CoV-2 todos negativos.
Se realizó una endoscopia digestiva alta en la que se observaron cordones varicosos con várices menores de 5 mm, sin estigmas de sangrado reciente (Paquet III), ni várices gástricas que, sumados a la ecogenicidad heterogénea hepática documentada en la ecografía abdominal, llevaron a considerar que presentaba cirrosis de origen alcohólico (consumo de riesgo de 88 g de alcohol, 8,7 unidades de bebida estándar por 10 años hasta noviembre de 2021), pero no había evidencia de sangrado portal hipertensivo; por tanto, era necesario descartar las causas no hepáticas de la encefalopatía.
Se realizó una punción lumbar que no tuvo anormalidades en el citoquímico, un estudio microbiológico para panel infeccioso molecular que resultó negativo para neuroinfección, y una citología y un inmunofenotipo no que presentaron sospecha de compromiso tumoral. Finalmente, se realizó una RMN nuclear en la que se observaron hiperintensidades en núcleos de la base, predominio caudado bilateral en secuencias T1 y recuperación de la inversión atenuada de fluido (FLAIR) sugestivas de degeneración hepatolenticular (Figura 1).
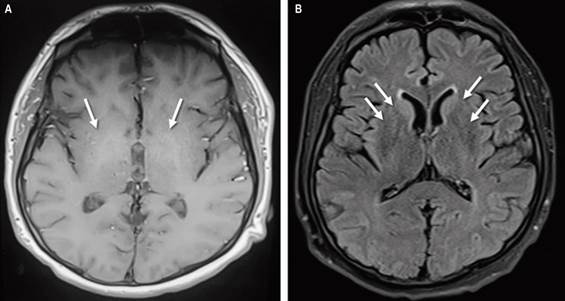
Fuente: archivo de los autores.
Figura 1 Resonancia magnética nuclear cerebral secuencias T1 (A) FLAIR (B). Las flechas muestran áreas de hiperintensidad en los núcleos basales bilaterales.
En la búsqueda de una enfermedad de Wilson, el área de oftalmología evaluó y descartó la presencia de anillos de Kayser-Fleischer, además se practicaron estudios de ceruloplasmina sérica (22 mg/dL) y cobre en orina (27,8 µg/24 horas), los cuales fueron normales.
Luego de 4 días de intubación se logró la extubación, 11 días después se dio egreso gracias a la mejoría de su condición neurológica, sin recurrencia de sangrado, hemoglobina estable y con plan de manejo ambulatorio con espironolactona, propranolol y recomendaciones nutricionales.
Discusión
La DHA es una entidad infrecuente; en un estudio retrospectivo con una población de más de 1000 pacientes con cirrosis hepática, solo 0,8% presentaba este diagnóstico y en otros reportes la prevalencia es aún más baja (menor al 0,5%)5-9, mientras que la etiología infecciosa viral es la más común10,11. El manganeso, causante de la DHA no wilsoniana, es el duodécimo elemento más importante, el quinto dentro de los metales12.
Varios sistemas enzimáticos dependen e interactúan con el manganeso, y es útil para la formación de cartílago, hueso y el mantenimiento de la mitocondria, además de la producción de glucosa13.
En estudios in vivo e in vitro, tanto en modelos animales como en humanos, se ha identificado que el SLC30A10 (Solute Carrier Family 30 Member 10), un transportador de salida del manganeso de la célula desempeña un papel vital en la regulación de los niveles de este metal. En individuos predispuestos con exposición ambiental debido a un sitio de residencia en sectores industriales, mineros o a su ocupación, las mutaciones que llevan a pérdida de función del SLC20A10 conducen a retención del manganeso en el cerebro, particularmente en los ganglios basales y el hígado, lo que genera neurotoxicidad y daño hepático, respectivamente14. Por otro lado, la enfermedad preexistente y la formación de derivaciones portosistémicas promueven una liberación de manganeso a la circulación sistémica y su depósito en el cerebro15. Recientemente se reportó una serie de cinco casos de DHA, cuatro de ellos tenían cirrosis y uno tenía hepatitis crónica por el virus de la hepatitis C y todos presentaban derivaciones portosistémicas16.
Las manifestaciones clínicas de la enfermedad son muy heterogéneas. La mayoría de los pacientes presentan cambios en el comportamiento, rasgos parkinsonianos y distonía con alteración de la marcha17. En el caso en mención, la presentación inicial fue un cambio en el comportamiento que alertó a sus familiares, luego una fase de agitación psicomotora durante la estancia en urgencias y posteriormente un deterioro del estado de conciencia que requirió intubación orotraqueal y ventilación mecánica invasiva en la unidad de cuidados intensivos (UCI). Estos cambios ocurren producto del incremento de manganeso en la circulación sistémica y su subyacente depósito cerebral, que puede ocurrir en la corteza, puntualmente en los ganglios de la base, lo que conlleva estrés oxidativo e induce neurotoxicidad por múltiples mecanismos como muerte de células dopaminérgicas y alteración en la transmisión mediada por ácido γ-aminobutírico (GABA), ambos estímulos inhibitorios; liberación de glutamato, el neurotransmisor excitatorio primario, y una molécula de señalización crítica; déficit de colina, necesario para la síntesis de la acetilcolina, y aumento de la acetilcolinesterasa, enzima que la degrada13,18. Además, los depósitos en el sistema nervioso central conducen a disfunción mitocondrial de los astrocitos a nivel de los receptores de benzodiacepinas19. Esto se ha demostrado en animales, en los que se observa que los niveles de manganeso afectan los astrocitos, con una disminución consecuente del trasporte de alta afinidad de glutamato y muerte neuronal mediada por estrés a través de la activación de las vías del óxido nítrico-monofosfato de guanosina cíclico18.
El diagnóstico para confirmar o descartar la DHA requiere de anamnesis, hallazgos al examen físico, análisis sanguíneo y una neuroimagen como la tomografía axial computarizada con contraste, y más usualmente la RMN, la cual permite excluir otros diagnósticos como hematomas cerebrales, enfermedad de pequeños vasos y lesiones ocupantes de espacio8. En la DHA, la RMN revela cambios en los ganglios de la base con hiperintensidad en T1, especialmente en el globo pálido, una proporción importante en pacientes cirróticos, relacionados con la gravedad de la enfermedad20. Los laboratorios en ocasiones pueden llegar a ser útiles, sin embargo, cabe mencionar que los niveles de manganeso en sangre y orina indican exposición reciente (horas a días), pero no existen biomarcadores de exposición acumulada al manganeso, ni biomarcadores pronósticos de sus efectos neurotóxicos; por tanto, no son diagnósticos.
En el caso de sospecha de enfermedad de Wilson se pueden solicitar niveles de celuroplasmina y amoniaco para descartarla, que en el paciente fueron negativos, que además de no tener la presencia de anillos de Kayser-Fleischer en el examen físico permitieron reforzar la sospecha diagnóstica, la cual se complementó con los hallazgos en la RMN que correspondían a depósitos de manganeso como agente causal de la encefalopatía19. De todas maneras, existe una buena correlación entre los niveles de manganeso en sangre y la hiperintensidad en T1 observada por la RMN, no necesariamente correlacionados con la función neurológica, por lo que en última instancia el diagnóstico se basa en la sospecha clínica y la exclusión de causas más frecuentes sin que una prueba diagnóstica confirme otra etiología16,20.
Según la evidencia reciente en EH West Haven 4, es muy raro encontrar niveles normales de amonio con su alta probabilidad preprueba; no obstante, independiente del resultado, en el cuadro agudo la prioridad es dar manejo a los potenciales contribuyentes como sangrado gastrointestinal, alteraciones hidroelectrolíticas e infecciones21. Una vez lograda la estabilidad clínica, si el amoniaco es normal en EH, en el algoritmo diagnóstico es indispensable evaluar las etiologías causantes de la inflamación sistémica o neuronal que llevaron a la activación de la microglía y a la encefalopatía22.
Para el caso que se reporta, el punto de partida fue una cirrosis hepática ya establecida con manejo endoscópico previo de várices esofágicas, por lo que requería descartarse el sangrado, pero que en esta ocasión no presentaban estigmas de sangrado reciente. Los cultivos para descartar causas infecciosas sistémicas y del sistema nervioso central fueron negativos, sin embargo, no se podía descartar una sepsis con cultivo negativo sensible a ceftriaxona (adquirida en la comunidad) dada la acidosis metabólica, la posibilidad de translocación bacteriana debido al sangrado gastrointestinal, los signos clínicos de respuesta clínica, reactantes de inflamación aguda elevados y un recuento de leucocitos de 8040/mm3, lo cual es superior al esperado en una hipertensión portal y marcador pronóstico de falla orgánica y mortalidad, como lo mostró el estudio CANONIC en insuficiencia hepática aguda sobre crónica. Por tanto, fue importante el manejo antibiótico y de soporte en este paciente.
Se evaluaron etiologías metabólicas relacionadas con enfermedades por depósito, obteniendo un perfil ferrocinético normal. Por otra parte, al no tener pruebas estandarizadas para medir los niveles de manganeso, el amonio normal y las hiperintensidades en los ganglios de la base por RMN cerebral fueron claves en el diagnóstico diferencial entre enfermedad de Wilson y DHA no wilsoniana, a favor de este último, teniendo en cuenta los niveles normales de ceruloplasmina sérica, al igual que el cobre sérico y urinario. En la Tabla 2 se encuentran las características distintivas de las dos enfermedades.
Tabla 2 Principales diferencias de la enfermedad de Wilson y la degeneración hepatolenticular no wilsoniana
| Enfermedad de Wilson | Degeneración hepatolenticular no wilsoniana |
|---|---|
| Patrón de herencia autosómica | Adquirida |
| Depósitos de cobre en el hígado y el cerebro | Depósitos de manganeso en el cerebro |
| Incapacidad de unión de la ceruplasmina y el cobre | Asociación con insuficiencia hepática, principalmente de origen etílico |
| Mutación del transportador ATP7B | Aumento del transporte de manganeso a nivel intestinal y de los hepatocitos |
| Clínica extrapiramidal de predominio no parkinsoniano | Predomino de parkinsonismo y síndrome extrapiramidal |
| Afección multisistémica | Hallazgos histopatológicos similares a la enfermedad de Wilson |
Adaptado de: Rebolledo-García D, et al. Med Int Méx. 2015;31:478-84.
Con respecto al tratamiento, incluso si se reconoce la DHA, este es limitado; puede pensarse en el uso de agonistas de la dopamina en vista de los síntomas similares al Parkinson, sin embargo, gran parte de los pacientes no tienen buena respuesta. La evidencia del uso de rifaximina en el manejo de la EH en pacientes con DHA es pequeña y proviene de series de casos. Como ocurrió en la paciente, lo más relevante es el tratamiento de soporte de la enfermedad hepática y medidas nutricionales para el manejo ambulatorio.
Previamente se reportó una serie de tres casos de degeneración hepatolenticular wilsoniana, y en solo uno de ellos se documentaron hallazgos en los ganglios basales en la secuencia FLAIR con presentaciones clínicas diferentes; mientras que ahora se expone este caso en el que definitivamente las imágenes por RMN fueron fundamentales para el diagnóstico final de degeneración hepatolenticular no wilsoniana23. Esta experiencia muestra que las distintas formas en las que puede manifestarse esta enfermedad hacen necesario un enfoque integral que incluya la clínica y los estudios diagnósticos disponibles según cada caso.
Conclusión
La DHA es una enfermedad rara como causa de encefalopatía en pacientes con enfermedad hepática, especialmente asociada con etilismo, que genera síntomas neurológicos debilitantes, producto de los depósitos de manganeso en los ganglios de la base. Para su diagnóstico es necesario descartar otras causas más frecuentes y evaluar las imágenes cerebrales en búsqueda de los hallazgos típicos en la secuencia T1; mientras se define la etiología, el manejo se debe enfocar en las medidas de soporte de la enfermedad hepática.
Agradecimientos
Ninguno declarado por los autores.
REFERENCIAS
1. Woerkom W. La cirrhose hepatique avec alterations dan les centres nerveux evoluant chez des sujets dage moyen. Nouvelle Iconographie de la Salpetrie. Clinique Maladies Systeme Nerveux. 1914;7:41-51. [ Links ]
2. Victor M, Adams R, Cole M. The acquired (non-Wilsonian) type of chronic hepatocerebral degeneration. Medicine (Baltimore). 1965;44(5):345-96. https://doi.org/10.1097/00005792-196509000-00001 [ Links ]
3. Weissenborn K Parkinsonism in patients with cirrhosis: coincidence or consequence? Liver Int. 2011;31(5):592-94. https://doi.org/10.1111/j.1478-3231.2011.02470.x [ Links ]
4. Butterworth RF. Parkinsonism in cirrhosis: pathogenesis and current therapeutic options. Metab Brain Dis. 2013;28(2):261-7. https://doi.org/10.1007/s11011-012-9341-7 [ Links ]
5. Jog MS, Lang AE. Chronic acquired hepatocerebral degeneration: case reports and new insights. Mov Disord. 1995;10(6):714-22. https://doi.org/10.1002/mds.870100604 [ Links ]
6. Basile A, Jones E. Ammonia and gabaergic neurotransmission: interrelated factors in the pathogenesis of hepatic encephalopathy. Hepatology. 1997;25(6):1303. https://doi.org/10.1002/hep.510250636 [ Links ]
7. Rose C, Butterworth R, Zayed J, Normandin L, Todd K, Michalak A, et al. Manganese deposition in basal ganglia structures results from both portal- systemic shunting and liver dysfunction. Gastroenterology. 1999;117(3):640-44. https://doi.org/10.1016/S0016-5085(99)70457-9 [ Links ]
8. Park N, Park J, Choi Y, Yoo CI, Lee CR, Lee H, et al. Whole blood manganese correlates with high signal intensities on T1-weighted MRI patients with liver cirrhosis. Neurotoxicology. 2003;24(6):909-15. https://doi.org/10.1016/S0161-813X(03)00111-6 [ Links ]
9. Burkhard PR, Delavelle J, Du Pasquier R, Spahr L. Chronic parkinsonism associated with cirrhosis. A distinct subset of acquired hepatocerebral degeneration. Arch Neurol. 2003;60(4):521-28. https://doi.org/10.1001/archneur.60.4.521 [ Links ]
10. Saporta M, Andre’ C, Bahia P, Chimelli L, Pais PH, Silva TS, et al. Acquired hepatocerebral degeneration without overt liver disease. Neurology. 2004;63(10):1981-82. https://doi.org/10.1212/01.WNL.0000144193.62505.E6 [ Links ]
11. Machado J, Gaspar S, Barbosa V, Bina J, Matos H. High intensity signal in basal ganglia on T1 weighted images: case report in Manson’s schistosomiasis with portal systemic encephalopathy. Arq Neuropsiquiatr. 1999;57(2A):306-10. https://doi.org/10.1590/S0004-282X1999000200024 [ Links ]
12. Nadaska G, Lesny J, Michalik I. Environmental aspect of manganese chemistry. HEJ. 2012. ENV-100702A:1-16. [ Links ]
13. Cowan DM, Fan Q, Zou Y, Shi X, Chen J, Aschner M, Rosenthal FS, et al. Manganese exposure among smelting workers: blood manganese-iron ratio as a novel tool for manganese exposure assessment. Biomarkers. 2009;14(1):3-16. https://doi.org/10.1080/13547500902730672 [ Links ]
14. Balachandran RC, Mukhopadhyay S, McBride D, Veevers J, Harrison FE, Aschner M, et al. Brain manganese and the balance between essential roles and neurotoxicity. J Biol Chem. 2020;295(19):6312-29. https://doi.org/10.1074/jbc.REV119.009453 [ Links ]
15. Rajoriya N, Brahmania M, Feld JJ. Implications of manganese in chronic acquired hepatocerebral degeneration. Ann Hepatol. 2019;18(1):274-78. https://doi.org/10.5604/01.3001.0012.7938 [ Links ]
16. Kalisky I, Maor Y, Goldstein L, Inbar Y, Ben-Ari Z. Acquired hepatocerebral degeneration a case series of a rare condition. Isr Med Assoc J. 2023;25(1):8-12. [ Links ]
17. Ferrara J, Jankovic J. Acquired hepatocerebral degeneration. J Neurol. 2009;256(3):320-32. https://doi.org/10.1007/s00415-009-0144-7 [ Links ]
18. Felipo V, Butterworth RF. Mitochondrial dysfunction in acute hyperammonemia. Neurochem Int. 2002;40(6):487-91. https://doi.org/10.1016/S0197-0186(01)00119-X [ Links ]
19. Yin Z, Aschner JL, dos Santos AP, Aschner M. Mitochondrial-dependent manganese neurotoxicity in rat primary astrocyte cultures. Brain Res. 2008;1203:1-11. https://doi.org/10.1016/j.brainres.2008.01.079 [ Links ]
20. Pujol A, Pujol J, Graus F, Rimola A, Peri J, Mercader JM, et al. Hyperintense globus pallidus on T1- weighted MRI in cirrhotic patients is associated with severity of liver failure. Neurology. 1993;43(1):65-9. https://doi.org/10.1212/WNL.43.1_Part_1.65 [ Links ]
21. Deutsch-Link S, Moon AM. The Ongoing Debate of Serum Ammonia Levels in Cirrhosis: the Good, the Bad, and the Ugly. Am J Gastroenterol. 2023;118(1):10-3. https://doi.org/10.14309/ajg.0000000000001966 [ Links ]
22. Butterworth RF. The liver-brain axis in liver failure: neuroinflammation and encephalopathy. Nat Rev Gastroenterol Hepatol. 2013;10(9):522-8. https://doi.org/10.1038/nrgastro.2013.99 [ Links ]
23. Castaño O, Gómez DM, Ocampo JM, Casanova ME. Degeneración hepatolenticular: a propósito de tres casos. 2017;30(4):436-42. https://doi.org/10.17533/udea.iatreia.v30n4a07 [ Links ]
Citación: Acosta-Pérez T, Rivera-Martínez WA, Galvis-Martínez C, Ocampo-Chaparro JM, Gómez-Ramírez DM. Manganeso asociado con degeneración hepatolenticular no wilsoniana como causa infrecuente de encefalopatía: reporte de caso. Revista. colomb. Gastroenterol. 2023;38(4):510-515. https://doi.org/10.22516/25007440.989
Recibido: 14 de Noviembre de 2022; Aprobado: 18 de Abril de 2023
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
