











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink
Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a systemic and immune-mediated condition associated with fibroinflammatory lesions that can occur in any organ1-4. It generally manifests in patients between 50 and 60 with a male/female preponderance of 2:11,3,5,6. The final diagnosis requires clinical and radiological evaluation, serum IgG4 levels, and characteristic histopathological findings; however, none of these approaches alone provide definitive evidence for accurate patient classification7. Treatment mainly involves systemic corticosteroids8, and remission rates are reported to be up to 70%, with annual relapse rates of 11.5%9. Other regimens, such as immunosuppressants in combination with corticosteroids and, recently, the anti-CD20 monoclonal antibody (rituximab), have demonstrated efficacy10,11.
Clinical case
We present the case of a 59-year-old male patient with a medical history of primary hypertension, heart failure, and diabetes mellitus, the latter appearing at 55 years of age. Ten months before hospital admission, he exhibited jaundice, choluria, abdominal pain in the right hypochondrium, asthenia, and pruritus. The liver profile revealed cholestasis (Table 1); viral hepatitis A, B, and C studies were negative. Abdominal CT showed dilation of the intrahepatic bile ducts, and the common bile duct dilated up to 12 mm with diffuse thickening of its walls. Two weeks before admission, the patient presented with nausea, and the abdominal pain intensified, coupled with a weight loss of 17 kg during the disease, which resulted in his admission due to suspicion of malignant neoplasm of the bile duct.
Table 1 Biochemical monitoring
| Ten months before | Nine months before | Emergency admission | Before starting corticosteroids | Seven days posttherapy | |
|---|---|---|---|---|---|
| TB mg/dL (NV: 0.2-1.3) | 11.4 | 4.27 | 1.17 | 0.55 | 0.5 |
| DB mg/dL (NV: 0-0.3) | 5.36 | 2.14 | 0.6 | 0.3 | 0.3 |
| IB mg/dL (NV: 0-1.1) | 6.94 | 2.13 | 0.5 | 0.2 | 0.2 |
| AP IU/L (NV: 38-126) | 1001 | 1042 | 356 | 315 | 176 |
| GGTP IU/L (NV: 15-73) | 310 | 413 | 240 | 154 | 109 |
| GOT IU/L (NV: 15-46) | 86 | 177 | 54 | 49 | 38 |
| GPT UI/L (VN: 15-35) | 69 | 102 | 34 | 29 | 33 |
AP: alkaline phosphatase; DB: direct bilirubin; GGTP: gamma-glutamyl transpeptidase; GOT: glutamic-oxaloacetic transaminase; GPT: glutamic-pyruvic transaminase; IB: indirect bilirubin; TB: total bilirubin. Prepared by the authors.
On physical examination, he was awake, thin, and anicteric, and his abdomen showed no significant findings. Ancillary tests showed leukocytes at 3,920 cells/mm3, hemoglobin of 12.7 gr/dL, platelets at 183,000/mm3, and a liver profile with cholestasis (Table 1). Kidney function, tumor markers (CEA, CA125, alpha-fetoprotein), and rheumatologic markers (ANA and ANCA) were negative. The examinations were complemented by imaging studies, which showed chronic inflammatory changes in the right submandibular gland and the absence of the left submandibular gland (Figure 1).
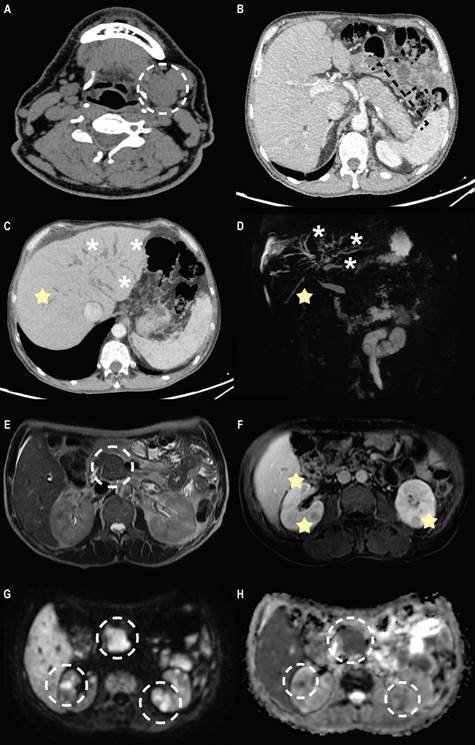
Source: Patient’s medical record.
Figure 1 A. Non-contrast computed tomography (CT) two years before admission shows increased volume of the left submandibular gland, associated with some adjacent lymphadenopathy. B. Contrast-enhanced CT shows a characteristic hypoattenuating halo around the body and tail of the pancreas, a finding suggestive of diffuse autoimmune pancreatitis. C and D. MinIP reconstruction and magnetic resonance cholangiopancreatography show dilation of the intrahepatic bile duct (asterisk) and segmental stricture areas (star). E. Axial T2-weighted magnetic resonance image shows well-demarcated focal enlargement at the head level, which correlates with diffusion restriction (G). F, G and H. The contrast-enhanced T1-weighted axial magnetic resonance image shows multiple well-defined renal nodular lesions, with enhancement upon contrast administration and correlation with diffusion restriction (G and H).
Due to this last finding, the anamnesis was expanded; the patient reported that two years ago, he underwent a left submandibulectomy for suspected neoplasia whose pathological study was compatible with Küttner’s tumor (Figure 2). The electrophoresis of proteins revealed an increase in total proteins and immunoglobulins, with an increase in IgG by 2,784 mg/dL (NV: 700-1690 mg/dL) and IgG4 by 556 (NV: 11-157 mg/dL). Proteinuria was 206.4 mg/24 hours, and the complement C3 level was decreased by 7 mg/dL (NV: 12.9-39.2).
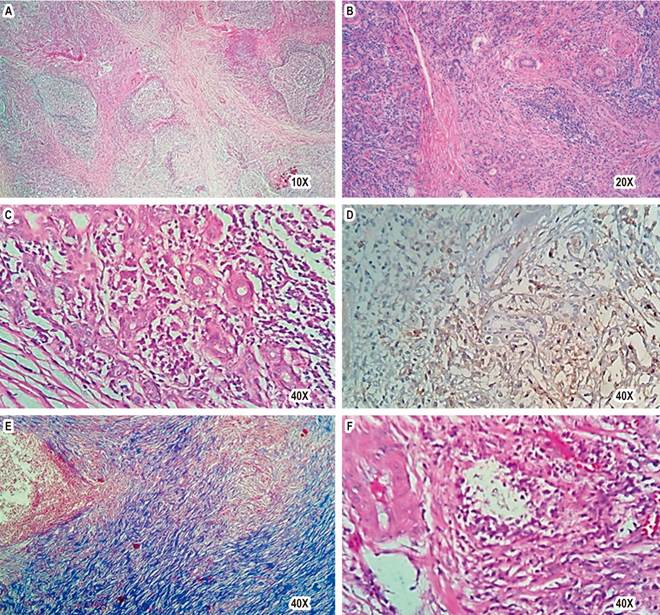
Source: Patient’s medical record.
Figure 2 Histopathological findings of IgG4-RD in the submandibular gland. A. Panoramic view of submandibular gland with lymphoplasmacytic inflammation and interstitial fibrosis (H. E.). B and C. Chronic inflammation: lymphocytes and plasma cells surrounding excretory ducts (H. E.). D. CD138-positive plasma cells. E. Storiform fibrosis with Masson staining. F. Phlebitis obliterans (H. E.).
Magnetic resonance cholangiopancreatography identified areas of stricture and dilation of the multifocal intrahepatic bile ducts and mural thickening of the common bile duct with stricture of its distal third, the prominent pancreas with a cephalic predominance, and the pancreatic duct with areas of stricture and nodular renal cortical lesions (Figure 1). The study was complemented by endoscopic ultrasonography demonstrating an 8 mm common bile duct with walls thickened by up to 3.5 mm, and the pancreas had a pseudotumoral appearance and scalloped edges.
Given the sum of the criteria, the diagnosis of IgG4-RD with multiorgan involvement (bile duct, pancreas, kidney, submandibular gland) was proposed. After a defocus study, treatment with prednisone 40 mg/day (0.8 mg/kg) was started. There were no complications during the first week of treatment, and he was discharged from the hospital. In the third week, the patient was asymptomatic with a reduction of prednisone to 30 mg/day, and leflunomide 20 mg/day was added to reduce the adverse effects of steroids and ensure remission of the disease.
Discussion
IgG4-RD was first recognized as a distinct disease in 200312: since then, it has been described more frequently by doctors from different specialties who participate in evaluating patients with this condition and, thus, a greater number of patients diagnosed worldwide7,13,14. It manifests as a multiorgan disease and can often be confused with diseases of infectious, autoimmune, or malignant origin7, just like the diagnosis made on admission of this case.
The classic patient is a middle-aged man (58.8 years)13, compared to the typical manifestation of predominantly female autoimmune diseases3. In autoimmune pancreatitis, the male/female ratio is 2:115; however, if other organs, such as the head or neck, are affected, the proportion is comparable16. The reason for the differential expression according to the involvement of other organs between both sexes is unclear3.
IgG4-RD follows a biphasic course17. In the inflammatory phase, polyclonal B and T cell subpopulations infiltrate affected tissues and undergo antigen-mediated interactions, releasing inflammatory and profibrotic cytokines18. T-follicular helper cells play an essential role in promoting the clonal expansion of IgG4-engaged B cells and enhancing the maturation of naïve B cells into mature plasma cells, with the production of IgG419. In the second fibrotic phase, innate immune cells, such as M2 macrophages, infiltrate IgG4-RD lesions and secrete profibrotic cytokines20. Activated fibroblasts deposit extracellular matrix, resulting in a dense stromal reaction that distorts tissue architecture, manifesting dysfunction and possibly organ failure17.
Four IgG4-RD phenotypes have been described: pancreato-hepato-biliary disease (31%), retroperitoneal fibrosis with or without aortitis (24%), head and neck-limited disease (24%), and classic Mikulicz syndrome with systemic involvement (22%)15.
In 2019. the American College of Rheumatology and the European Alliance of Associations for Rheumatology (ACR/EULAR) presented the IgG4-RD classification criteria. These were developed based on a three-step classification process: an entry criterion, a set of exclusion criteria, and weighted inclusion criteria7,21.
The initial clinical manifestation of this case was the involvement of the submandibular gland. In this regard, one in five patients with IgG4-RD in the head and neck region has the disease that originates in one or more salivary glands1. In decreasing order of prevalence, the submandibular gland, parotid gland, sublingual gland, and minor salivary glands are included1. The most common salivary gland disease associated with IgG4-RD is chronic sclerosing sialadenitis or Küttner’s tumor. This chronic benign inflammatory disorder most frequently affects the submandibular glands22. It appears as an increase in size or pseudotumor lesion in the region of the affected salivary gland and is usually unilateral; however, bilateral cases have been described23. The history and clinical findings are often highly suggestive of neoplasia and, as such, should always remain at the top of the list of differential diagnoses, considering IgG4-RD as a diagnosis of exclusion4.
Meanwhile, during the evolution of his disease, the patient had biliary and pancreatic involvement. In this context, two subtypes of autoimmune pancreatitis (AIP) are known, of which only one (Type 1) is associated with IgG4-RD24,25. Type 1 AIP, the most common form, is characterized by the classic histopathological findings of lymphoplasmacytic sclerosing pancreatitis24.
Clinically, it manifests as obstructive jaundice (generally painless) associated with weight loss and fatigue (simulating pancreatic neoplasia) or acute pancreatitis (generally benign without necrosis)25. CT features include diffuse pancreatic enlargement with late enhancement and a low-density capsule-like ring. Diffuse and irregular narrowing of the main pancreatic duct on magnetic resonance cholangiopancreatography is also definite for AIP26,27.
IgG4-related sclerosing cholangitis (IgG4-SC) has a cholangiographic appearance similar to primary sclerosing cholangitis (PSC). Both IgG4-SC and AIP respond well to steroid therapy28,29. On the contrary, PSC is progressive, resistant to treatment, affects both the intrahepatic and extrahepatic bile ducts, and causes biliary cirrhosis30. Another differential diagnosis to consider in this context is hilar cholangiocarcinoma, which often requires a transpapillary endoscopic biopsy30 because neither serum IgG4 concentrations nor cholangiographic or cholangioscopic findings differentiate these disorders clearly but may suggest a particular approach31,32.
In this case, imaging studies revealed characteristic renal lesions indicating involvement of this organ. Tubulointerstitial nephritis has been described as the most common form of IgG4-RD in the kidneys33. The critical difference between kidney involvement and other solid organ involvement is the low complement concentrations. It is not well understood but is not believed to be related to IgG4 because this molecule does not bind to complement. This phenomenon is not replicated to a large extent in most other organs involving IgG4-RD33. Clinically, these patients may experience kidney failure and even end-stage renal disease. Although proteinuria may develop, it is often in the subnephrotic range. Imaging studies will indicate significantly enlarged kidneys and hypodense lesions evident on CT images and non-enhancing in post-contrast studies34. All these findings were apparent in this case.
A key point to consider is serum IgG4 concentrations, initially a critical characteristic for diagnosing IgG4-RD but described as neither necessary nor sufficient26. A cohort of patients with IgG4-RD demonstrated that high levels of IgG4 do not confirm the diagnosis, and low serum levels do not exclude it35.
Regarding treating IgG4-RD, it will depend on the severity of the disease, the affected organs, and the individual factors of each patient36.
Glucocorticoids are the cornerstone of treatment in patients with active IgG4-RD both at onset and at relapse. The goal is to resolve symptoms and normalize biochemistry and radiological findings. Generally, improvement should occur in days to several weeks, depending on the organs involved37; however, the lack of response to glucocorticoid therapy suggests that the diagnosis is incorrect and should be reconsidered36. The general practice is to start with a dose between 0.6-1 mg/kg/day38. Evidence suggests that the initial dose should be maintained for 2 to 4 weeks and then gradually reduced by 5 mg every two weeks throughout 4 to 6 weeks6. During this time, the patient should be monitored for common disease relapses, especially at lower doses and after discontinuation39.
The role of immunomodulators in the induction of IgG4-RD remission has not been established37. Current guidelines indicate that some, but not all, patients require the combination of glucocorticoids and an immunosuppressant from the start of treatment; this is because monotherapy with glucocorticoids will ultimately fail to control the disease, and long-term toxicity represents a high risk for patients11. Besides, biological agents such as rituximab (RTX) can be combined with corticosteroids to induce disease remission and allow early tapering of steroids10. It has also been used successfully in patients who showed resistance or side effects to classic treatments (steroids/immunomodulators)11. In maintenance therapy, RTX is superior to other regimens in reducing the relapse rate; however, dosing protocols are not established in this setting40.
In this case, a new biopsy sample was rejected because there was previously a pathological finding within the IgG4-RD spectrum (Küttner’s tumor). Previously, the diagnosis was obtained incidentally from extensive surgical specimens resected for suspected malignancy. Nonetheless, given the greater recognition of this condition, the current diagnosis is made using increasingly smaller biopsy samples and from accessible sites, even in selected cases, with the addition of classic non-invasive criteria and no anatomopathological study7,41.

