










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista colombiana de Gastroenterología
versão impressa ISSN 0120-9957versão On-line ISSN 2500-7440
Rev. colomb. Gastroenterol. vol.38 no.4 Bogotá out./dez. 2023 Epub 26-Fev-2024
https://doi.org/10.22516/25007440.978
Reporte de casos
Enfermedad relacionada con inmunoglobulina G4, un diagnóstico a tener en cuenta: a propósito de un caso
1
*
 http://orcid.org/0000-0002-8416-1666
http://orcid.org/0000-0002-8416-1666
1
http://orcid.org/0000-0003-2623-0296
2
http://orcid.org/0000-0002-3620-1918
1 Médico gastroenterólogo, asistente en el servicio de gastroenterología, Hospital Nacional Guillermo Almenara Irigoyen. Lima, Perú.
2 Médica asistente en el servicio de radiología, Hospital Nacional Guillermo Almenara Irigoyen. Lima Perú.
3 Médico residente en el servicio de gastroenterología, Hospital Nacional Guillermo Almenara Irigoyen. Lima, Perú.
4 Médica asistente en el servicio de anatomía patológica, Hospital Nacional Guillermo Almenara Irigoyen. Lima, Perú.
La enfermedad relacionada con inmunoglobulina G4 (ER-IgG4) es una condición inmunomediada y multisistémica asociada al desarrollo de lesiones fibroinflamatorias en cualquier órgano. El diagnóstico se realiza bajo la suma de criterios clínicos, serológicos, radiológicos e histopatológicos; sin embargo, este suele ser difícil debido a su similitud con neoplasias, infecciones u otras enfermedades inmunomediadas. El tratamiento se basa en corticosteroides, en una posible combinación con inmunomoduladores. El presente caso trata de un varón de 59 años con historia de síndrome ictérico y baja de peso, admitido por sospecha de neoplasia maligna de la vía biliar. Las imágenes revelaron estenosis con dilatación de las vías biliares intrahepáticas, páncreas prominente, estenosis del conducto pancreático y lesiones nodulares renales. Debido al antecedente de submandibulectomía izquierda 2 años antes de la enfermedad actual e histología compatible con tumor de Küttner, más los valores elevados de IgG4 sérico, se estableció el diagnóstico de ER-IgG4. Inició tratamiento con corticosteroides y cursó asintomático durante el seguimiento.
Palabras claves: Enfermedad relacionada con IgG4; pancreatitis autoinmune; inmunoglobulina G4
Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated and multisystemic condition associated with developing fibroinflammatory lesions in any organ. The diagnosis is made based on the sum of clinical, serological, radiological, and histopathological criteria; however, this is often difficult due to its similarity to neoplasms, infections, or other immune-mediated diseases. Treatment is based on corticosteroids, in a possible combination with immunomodulators. The present case concerns a 59-year-old man with a history of jaundice syndrome and weight loss, admitted for suspected malignant neoplasia of the bile duct. Imaging revealed stricture with dilation of the intrahepatic bile ducts, prominent pancreas, pancreatic duct stricture, and nodular renal lesions. Due to the history of left submandibulectomy two years before the current disease and histology compatible with Küttner’s tumor, plus the high IgG4 serum values, the diagnosis of IgG4-RD was established. He started treatment with corticosteroids and was asymptomatic during follow-up.
Keywords: IgG4-related disease; autoimmune pancreatitis; immunoglobulin G4
Introducción
La enfermedad relacionada con inmunoglobulina G4 (ER-IgG4) es una condición sistémica e inmunomediada asociada con lesiones fibroinflamatorias que pueden ocurrir en cualquier órgano1-4. Generalmente se presenta en pacientes entre los 50 y 60 años con una preponderancia de sexo masculino/femenino de 2:11,3,5,6. El diagnóstico definitivo requiere evaluación clínica, radiológica, niveles séricos de IgG4 y hallazgos histopatológicos característicos; no obstante, ninguno de estos enfoques por sí solo proporciona evidencia definitiva para la clasificación precisa de los pacientes7. El tratamiento es principalmente con corticosteroides sistémicos8, y se reportan tasas de remisión hasta en el 70%, con tasas de recaída anual del 11,5%9. Otros regímenes como los inmunosupresores en combinación con los corticosteroides y recientemente el anticuerpo monoclonal anti-CD20 (rituximab) han demostrado eficacia10,11.
Caso clínico
Paciente de sexo masculino de 59 años, con antecedentes médicos de hipertensión arterial esencial, insuficiencia cardíaca y diabetes mellitus, este último con debut a los 55 años. Diez meses antes de su ingreso hospitalario, presentó ictericia, coluria, dolor abdominal en el hipocondrio derecho, astenia y prurito. El perfil hepático reveló colestasis (Tabla 1); los estudios de hepatitis viral A, B y C fueron negativos; la tomografía abdominal mostró una dilatación de las vías biliares intrahepáticas y el colédoco dilatado hasta en 12 mm con engrosamiento difuso de sus paredes. Dos semanas antes de la admisión, el paciente presenta náuseas y el dolor abdominal se intensifica, sumado a una pérdida ponderal de 17 kg durante el curso de la enfermedad, lo que motivó su ingreso ante la sospecha de neoplasia maligna de la vía biliar.
Tabla 1 Seguimiento bioquímico
| 10 meses antes | 9 meses antes | Ingreso a emergencias | Previo al inicio de corticoides | 7 días posterapia | |
|---|---|---|---|---|---|
| BT mg/dL (VN: 0,2-1,3) | 11,4 | 4,27 | 1,17 | 0,55 | 0,5 |
| BD mg/dL (VN: 0-0,3) | 5,36 | 2,14 | 0,6 | 0,3 | 0,3 |
| BI mg/dL (VN: 0-1,1) | 6,94 | 2,13 | 0,5 | 0,2 | 0,2 |
| FA UI/L (VN: 38-126) | 1001 | 1042 | 356 | 315 | 176 |
| GGTP UI/L (VN: 15-73) | 310 | 413 | 240 | 154 | 109 |
| TGO UI/L (VN: 15-46) | 86 | 177 | 54 | 49 | 38 |
| TGP UI/L (VN: 15-35) | 69 | 102 | 34 | 29 | 33 |
BD: bilirrubina directa; BI: bilirrubina indirecta; BT: bilirrubina total; FA: fosfatasa alcalina; GGTP: gamma-glutamil transpeptidasa; TGO: transaminasa glutámico-oxalacética; TGP: transaminasa glutámico-pirúvica. Tabla elaborada por los autores.
En la exploración física, se encontró despierto, adelgazado, anictérico y el abdomen lució sin hallazgos significativos. Los exámenes auxiliares mostraron leucocitos en 3920 cel/mm3, hemoglobina 12,7 gr/dL, plaquetas 183.000/mm3, perfil hepático con colestasis (Tabla 1). La función renal, los marcadores tumorales (CEA, CA-125, alfafetoproteína) y los marcadores reumatológicos ANA y ANCA fueron negativos. Los exámenes se complementaron con estudios de imagen, que evidenciaron cambios inflamatorios crónicos en la glándula submaxilar derecha y ausencia de la glándula submandibular izquierda (Figura 1).

Fuente: expediente médico del paciente.
Figura 1 A. La tomografía axial computarizada (TAC) sin contraste 2 años antes de la admisión muestra un aumento de volumen de la glándula submandibular izquierda, asociado con algunas adenopatías adyacentes. B. La TAC con contraste muestra un halo hipoatenuante característico alrededor del cuerpo y la cola del páncreas, un hallazgo sugestivo de pancreatitis autoinmune difusa. C y D. La reconstrucción MinIP y la colangiorresonancia muestran una dilatación de la vía biliar intrahepática (asterisco), así como áreas de estenosis segmentarias (estrella). E. Imagen de resonancia magnética axial ponderada en T2 que muestra un agrandamiento focal bien delimitado a nivel de la cabeza, que se correlaciona con restricción en la difusión (G). F, G y H. Imagen de resonancia magnética axial ponderada en T1 con contraste que muestra múltiples lesiones nodulares renales bien definidas, las cuales muestran realce a la administración de contraste y se correlacionan con restricción a la difusión (G y H).
Debido a este último hallazgo se amplió la anamnesis, en la que el paciente reportó que hace 2 años se le realizó una submandibulectomía izquierda por sospecha de neoplasia cuyo estudio anatomopatológico resultó compatible con un tumor de Küttner (Figura 2). El proteinograma electroforético reveló una elevación de proteínas totales, así como de inmunoglobulinas, con un aumento de la IgG en 2784 mg/dL (VN: 700-1690 mg/dL) y de la IgG4 en 556 (VN: 11-157 mg/dL). La proteinuria en 24 horas fue de 206,4 mg/24 horas, y el nivel de complemento C3 se mostró disminuido en 7 mg/dL (VN: 12,9-39,2).
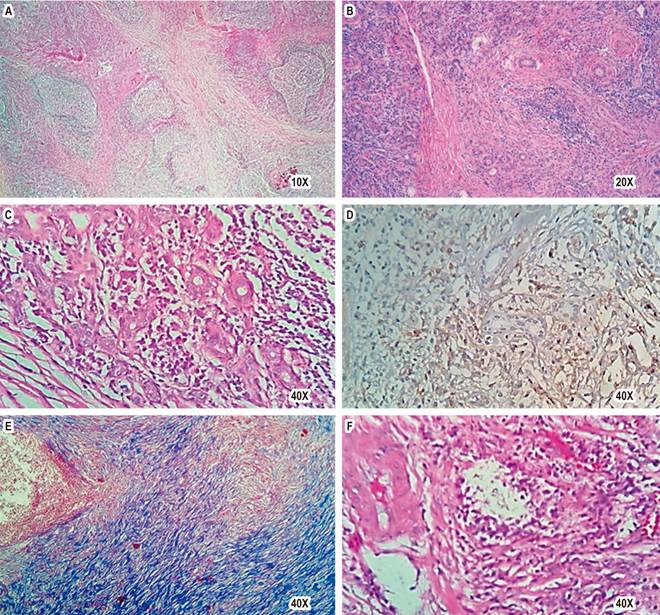
Fuente: expediente médico del paciente.
Figura 2 Hallazgos histopatológicos de ER-IgG4 en la glándula submaxilar. A. Vista panorámica de glándula submaxilar con inflamación linfoplasmocitaria y fibrosis intersticial (H. E.). B y C. Inflamación crónica: linfocitos y células plasmáticas rodeando conductos excretores (H. E.). D. Células plasmáticas positivas para CD138. E. Fibrosis estoriforme con coloración de Masson. F. Flebitis obliterante (H. E.).
La colangiopancreatografía por resonancia magnética identificó zonas de estenosis y dilatación de las vías biliares intrahepáticas multifocales y engrosamiento mural del conducto biliar común con estenosis de su tercio distal; el páncreas prominente a predominio cefálico, el conducto pancreático con áreas de estenosis y lesiones nodulares renales corticales (Figura 1). El estudio se complementa con una ultrasonografía endoscópica que demostró un colédoco de 8 mm con paredes engrosadas hasta en 3,5 mm, y el páncreas tiene un aspecto pseudotumoral y bordes festoneados.
Ante la suma de criterios, se planteó el diagnóstico de ER-IgG4 con compromiso multiorgánico (vía biliar, páncreas, riñón, glándula submandibular). Previo estudio de desfocalización, se inició el tratamiento con prednisona de 40 mg/día (0,8 mg/kg). En la evolución no presentó intercurrencias durante la primera semana de tratamiento y recibió el alta hospitalaria. En la tercera semana el paciente estuvo asintomático con una reducción de prednisona a 30 mg/día y se agregó leflunomida de 20 mg/día; este último con la finalidad de disminuir los efectos adversos de los esteroides y asegurar la remisión de la enfermedad.
Discusión
La ER-IgG4 fue reconocida por primera vez como una enfermedad distinta en 200312; desde entonces se describe con mayor frecuencia por médicos de diferentes especialidades que participan en la evaluación de pacientes con esta afección y, con esto, un mayor número de pacientes diagnosticados en todo el mundo7,13,14. Se presenta como una enfermedad multiorgánica y puede confundirse a menudo con enfermedades de origen infeccioso, autoinmune o maligno7; como el diagnóstico que se planteó en la admisión del presente caso.
El paciente clásico es un varón de mediana edad (58,8 años)13, en contraste con la presentación típica de las enfermedades autoinmunes de predominio femenino3. En la pancreatitis autoinmune, la relación masculino/femenino es 2:115; no obstante, si se afectan otros órganos como la cabeza o el cuello, la proporción es equiparable16. La razón de la expresión diferencial según el compromiso de otros órganos entre ambos sexos no está clara3.
La ER-IgG4 sigue un curso bifásico17. En la fase inflamatoria, las subpoblaciones de células B y T policlonales se infiltran en los tejidos afectados y experimentan interacciones mediadas por antígenos, lo que conduce a la liberación de citocinas inflamatorias y profibróticas18. Las células T helper foliculares desempeñan un papel importante al promover la expansión clonal de células B comprometida con IgG4 y mejorar la maduración de las células B naïve en células plasmáticas maduras, con la producción de IgG419. En la segunda fase fibrótica, las células inmunitarias innatas, como los macrófagos M2, se infiltran en las lesiones de la ER-IgG4 y secretan citocinas profibróticas20. Los fibroblastos activados depositan matriz extracelular, lo que da como resultado una reacción estromal densa que distorsiona la arquitectura del tejido que se manifiesta en disfunción y posiblemente fallo del órgano17.
Se han descrito 4 fenotipos de la ER-IgG4: enfermedad pancreato-hepatobiliar (31%), fibrosis retroperitoneal con o sin aortitis (24%), enfermedad limitada de cabeza y cuello (24%) y el síndrome de Mikulicz clásico con compromiso sistémico (22%)15.
En 2019, el Colegio Americano de Reumatología y la Liga Europea contra las Enfermedades Reumáticas (ACR/EULAR) presentaron los criterios de clasificación de la ER-IgG4. Estos se desarrollaron con base en un proceso de clasificación de tres pasos, que consiste en un criterio de entrada, un conjunto de criterios de exclusión y criterios de inclusión ponderados7,21.
La presentación clínica inicial de este caso fue la afectación de la glándula submandibular. Al respecto, uno de cada cinco pacientes que presentan una ER-IgG4 en la región de la cabeza y el cuello tiene la enfermedad que se origina en una o más de las glándulas salivales1. En orden decreciente de prevalencia se incluyen la glándula submandibular, la glándula parótida, la glándula sublingual y las glándulas salivales menores1. La enfermedad de las glándulas salivales más común asociada con ER-IgG4 es la sialoadenitis esclerosante crónica o tumor de Küttner. Este es un trastorno inflamatorio benigno crónico que afecta con mayor frecuencia a las glándulas submandibulares22, y se presenta como un aumento de tamaño o lesión pseudotumoral en la región de la glándula salival afectada y suele ser unilateral; sin embargo, se han descrito casos bilaterales23. La historia y los hallazgos clínicos a menudo son muy sugestivos de neoplasia y, como tal, siempre debe permanecer en la parte superior de la lista de diagnósticos diferenciales, teniendo en cuenta a la ER-IgG4 como un diagnóstico de exclusión4.
Por otra parte, durante la evolución de su enfermedad el paciente presentó compromiso biliar y pancreático. En este contexto, se conocen dos subtipos de pancreatitis autoinmune (PAI), de los cuales solo uno (tipo 1) está asociado con ER-IgG424,25. La PAI tipo 1, la forma más común, se caracteriza por los hallazgos histopatológicos clásicos de pancreatitis esclerosante linfoplasmocítica24.
Clínicamente se presenta como ictericia obstructiva (generalmente indolora) asociada con pérdida de peso y fatiga (simulando una neoplasia de páncreas) o pancreatitis aguda (generalmente benigna sin necrosis)25. Las características tomográficas incluyen un agrandamiento pancreático difuso con realce tardío y un anillo de baja densidad similar a una cápsula. El estrechamiento difuso e irregular del conducto pancreático principal en la colangiopancreatografía por resonancia magnética también es altamente específico para la PAI26,27.
La colangitis esclerosante relacionada con IgG4 (CE-IgG4) tiene una apariencia colangiográfica similar a la de la colangitis esclerosante primaria (CEP). Tanto la CE-IgG4 como la PAI responden bien a la terapia con esteroides28,29. Por el contrario, la CEP es progresiva, resistente al tratamiento, afectan tanto a las vías biliares intrahepáticas como extrahepáticas y provoca cirrosis biliar30. Por otra parte, un diagnóstico diferencial adicional para tener cuenta en este contexto es el colangiocarcinoma hiliar, el cual a menudo requiere de una biopsia endoscópica transpapilar30 debido a que ni las concentraciones séricas de IgG4 ni los hallazgos colangiográficos o colangioscópicos diferencian claramente estos trastornos, aunque pueden sugerir cierta aproximación31,32.
En este caso, los estudios de imágenes revelaron lesiones renales características que indicaban compromiso de este órgano. Se ha descrito a la nefritis tubulointersticial como la forma más común de ER-IgG4 en los riñones33. La diferencia clave entre la afectación renal y otra afectación de órganos sólidos es la presencia de bajas concentraciones del complemento, que aún no se conoce bien, pero no se cree que está relacionado con la IgG4 en sí misma porque esta molécula no se une al complemento y este fenómeno no se replica en gran medida en la mayoría de los otros órganos que involucran ER-IgG433. Clínicamente, estos pacientes pueden experimentar disfunción renal e incluso enfermedad renal en etapa terminal. Aunque puede desarrollarse proteinuria, a menudo es en rango subnefrótico. Los estudios de imágenes indicarán riñones significativamente agrandados y lesiones hipodensas evidentes en las imágenes de tomografía y que no realzan en los estudios poscontraste34. Todos estos hallazgos fueron evidenciados en este caso.
Un punto importante a tener en cuenta son las concentraciones séricas de IgG4, que inicialmente fue una característica clave para el diagnóstico de ER-IgG4, pero se ha descrito que no es necesario ni suficiente26. Una cohorte de pacientes con ER-IgG4 demostró que los niveles elevados de IgG4 no confirman el diagnóstico y los niveles séricos bajos no la excluyen35.
Respecto al tratamiento de la ER-IgG4, este dependerá de la gravedad de la enfermedad, los órganos afectados y factores individuales de cada paciente36.
Los glucocorticoides son la piedra angular del tratamiento en pacientes con ER-IgG4 activa tanto al inicio como en la recaída. El objetivo es resolver los síntomas, normalizar la bioquímica y los hallazgos radiológicos. Por lo general, la mejoría debe observarse en días a varias semanas, dependiendo de los órganos involucrados37; no obstante, la falta de respuesta a la terapia con glucocorticoides sugiere que el diagnóstico es incorrecto y debería replantearse36. La práctica general es iniciar con una dosis entre 0,6-1 mg/kg/día38. La evidencia sugiere que la dosis inicial debe mantenerse durante 2 a 4 semanas y luego reducirse gradualmente en 5 mg cada 2 semanas durante un período de 4 a 6 semanas6. Durante este tiempo, se debe controlar al paciente por recaídas de la enfermedad que son comunes, especialmente a dosis más bajas y luego de la interrupción39.
El papel de los inmunomoduladores en la inducción de la remisión de ER-IgG4 no se ha establecido claramente37. Las directrices actuales indican que algunos, pero no todos los pacientes requieren la combinación de glucocorticoides y un inmunosupresor desde el inicio del tratamiento; esto se debe a que la monoterapia con glucocorticoides finalmente no logrará controlar la enfermedad, y la toxicidad a largo plazo representan un alto riesgo para los pacientes11. Por otro lado, agentes biológicos como rituximab (RTX) se pueden usar en combinación con corticosteroides para inducir la remisión de la enfermedad y permitir la disminución temprana de esteroides10. De la misma manera, también se ha utilizado con éxito en pacientes que mostraron resistencia o efectos secundarios ante los tratamientos clásicos (esteroides/inmunomoduladores)11. En terapia de mantenimiento, RTX ha demostrado ser superior a los otros regímenes en la reducción de la tasa de recaídas; sin embargo, sus protocolos de dosificación no están claramente establecidos en este entorno40.
Para este caso, se desestimó realizar una nueva muestra de biopsia debido a que se contaba previamente con un hallazgo patológico dentro del espectro de la ER-IgG4 (tumor de Küttner). Anteriormente, el diagnóstico se obtenía de manera incidental a partir de extensas piezas quirúrgicas resecadas por sospecha de malignidad. No obstante, dado el mayor reconocimiento de esta afección, el diagnóstico actual se realiza utilizando muestras de biopsias cada vez más pequeñas y de sitios accesibles, incluso en casos seleccionados, bajo la suma de criterios clásicos no invasivos y sin requerir un estudio anatomopatológico7,41.
REFERENCIAS
1. Stone JH, Zen Y, Deshpande V. Mechanisms of disease: IgG4-related disease. N Engl J Med. 2012;366(6):539-51. https://doi.org/10.1056/NEJMRA1104650 [ Links ]
2. Ruiz-Rebollo ML, Álvarez-Quiñones-Sanz M, Fuertes-Alija JJ, Izquierdo-Santervás S. IgG4-related disease mimicking pancreatic tumour. Gastroenterología y Hepatología (English Edition). 2021;44(9):660-1. https://doi.org/10.1016/J.GASTRE.2020.07.026 [ Links ]
3. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385(9976):1460-71. https://doi.org/10.1016/S0140-6736(14)60720-0 [ Links ]
4. Johnston J, Allen JE. IgG4-related disease in the head and neck. Curr Opin Otolaryngol Head Neck Surg. 2018;26(6):403-8. https://doi.org/10.1097/MOO.0000000000000487 [ Links ]
5. Skillington SA, Ogden MA. IgG4-Related Disease and the Salivary Glands: A Review of Pathophysiology, Diagnosis, and Management. Otolaryngol Clin North Am. 2021;54(3):497-508. https://doi.org/10.1016/J.OTC.2021.02.002 [ Links ]
6. Wallace ZS, Perugino C, Matza M, Deshpande V, Sharma A, Stone JH. IgG4-Related Disease. Clin Chest Med. 2019;40(3):583-97. https://doi.org/10.1016/J.CCM.2019.05.005 [ Links ]
7. Wallace ZS, Naden RP, Chari S, Choi H, Della-Torre E, Dicaire JF, et al. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Arthritis Rheumatol. 2020;72(1):7-19. https://doi.org/10.1002/ART.41120 [ Links ]
8. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67(7):1688-99. https://doi.org/10.1002/ART.39132 [ Links ]
9. Yamamoto M, Yajima H, Takahashi H, Yokoyama Y, Ishigami K, Shimizu Y, et al. Everyday clinical practice in IgG4-related dacryoadenitis and/or sialadenitis: Results from the SMART database. Mod Rheumatol. 2015;25(2):199-204. https://doi.org/10.3109/14397595.2014.950036 [ Links ]
10. Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: Lessons from 10 consecutive patients. Medicine. 2012;91(1):57-66. https://doi.org/10.1097/MD.0B013E3182431EF6 [ Links ]
11. Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171-7. https://doi.org/10.1136/ANNRHEUMDIS-2014-206605 [ Links ]
12. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982-4. https://doi.org/10.1007/S00535-003-1175-Y [ Links ]
13. Uchida K, Masamune A, Shimosegawa T, Okazaki K. Prevalence of IgG4-Related Disease in Japan Based on Nationwide Survey in 2009. Int J Rheumatol. 2012;2012:358371. https://doi.org/10.1155/2012/358371 [ Links ]
14. Masamune A, Kikuta K, Hamada S, Tsuji I, Takeyama Y, Shimosegawa T, et al. Nationwide epidemiological survey of autoimmune pancreatitis in Japan in 2016. J Gastroenterol. 2020;55(4):462-70. https://doi.org/10.1007/S00535-019-01658-7 [ Links ]
15. Wallace ZS, Zhang Y, Perugino CA, Naden R, Choi HK, Stone JH. Clinical phenotypes of IgG4-related disease: an analysis of two international cross-sectional cohorts. Ann Rheum Dis. 2019;78(3):406-12. https://doi.org/10.1136/ANNRHEUMDIS-2018-214603 [ Links ]
16. Zen Y, Nakanuma Y. IgG4-related disease: A cross-sectional study of 114 cases. Am J Surg Pathol, 2010;34(12):1812-9. https://doi.org/10.1097/PAS.0B013E3181F7266B [ Links ]
17. Pillai S, Perugino C, Kaneko N. Immune mechanisms of fibrosis and inflammation in IgG4-related disease. Curr Opin Rheumatol. 2020;32(2):146-51. https://doi.org/10.1097/BOR.0000000000000686 [ Links ]
18. Della-Torre E, Feeney E, Deshpande V, Mattoo H, Mahajan V, Kulikova M, et al. B-cell depletion attenuates serological biomarkers of fibrosis and myofibroblast activation in IgG4-related disease. Ann Rheum Dis. 2015;74(12):2236. https://doi.org/10.1136/ANNRHEUMDIS-2014-205799 [ Links ]
19. Chen Y, Lin W, Yang H, Wang M, Zhang P, Feng R, et al. Aberrant Expansion and Function of Follicular Helper T Cell Subsets in IgG4-Related Disease. Arthritis Rheumatol. 2018;70(11):1853-65. https://doi.org/10.1002/ART.40556 [ Links ]
20. Furukawa S, Moriyama M, Tanaka A, Maehara T, Tsuboi H, Iizuka M, et al. Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Clinical Immunology. 2015;156(1):9-18. https://doi.org/10.1016/J.CLIM.2014.10.008 [ Links ]
21. Lanzillotta M, Lanzillotta M, Mancuso G, Mancuso G, Della-Torre E. Advances in the diagnosis and management of IgG4 related disease. BMJ. 2020;369:m1067. https://doi.org/10.1136/BMJ.M1067 [ Links ]
22. Bhatti RM, Stelow EB. IgG4-related disease of the head and neck. Adv Anat Pathol. 2013;20(1):10-6. https://doi.org/10.1097/PAP.0B013E31827B619E [ Links ]
23. Mulholland GB, Jeffery CC, Satija P, Côté DWJ. Immunoglobulin G4-related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg.2015;44(1):24. https://doi.org/10.1186/S40463-015-0071-9 [ Links ]
24. Chari ST, Kloeppel G, Zhang L, Notohara K, Lerch MM, Shimosegawa T. Histopathologic and clinical subtypes of autoimmune pancreatitis: The honolulu consensus document. Pancreas. 2010;39(5):549-54. https://doi.org/10.1097/MPA.0B013E3181E4D9E5 [ Links ]
25. Sah RP, Chari ST, Pannala R, Sugumar A, Clain JE, Levy MJ, et al. Differences in Clinical Profile and Relapse Rate of Type 1 Versus Type 2 Autoimmune Pancreatitis. Gastroenterology. 2010;139(1):140-8. https://doi.org/10.1053/J.GASTRO.2010.03.054 [ Links ]
26. Vashi B, Khosroshahi A. IgG4-Related Disease with Emphasis on Its Gastrointestinal Manifestation. Gastroenterol Clin North Am. 2019;48(2):291-305. https://doi.org/10.1016/j.gtc.2019.02.008 [ Links ]
27. Zheng Y, Elsayes KM, Waranch C, Abdelaziz A, Menias CO, Sandrasegaran K, et al. IgG4-related disease in the abdomen and pelvis: atypical findings, pitfalls, and mimics. Abdominal Radiology. 2020;45(8):2485-99. https://doi.org/10.1007/S00261-020-02526-2 [ Links ]
28. Erkelens GW, Vleggaar FP, Lesterhuis W, van Buuren HR, van der Werf SDJ. Sclerosing pancreato-cholangitis responsive to steroid therapy. Lancet. 1999;354(9172):43-4. https://doi.org/10.1016/S0140-6736(99)00603-0 [ Links ]
29. Smal AJ, Loftus CG, Smyrk TC, Baron TH. A case of IgG4-associated cholangitis and autoimmune pancreatitis responsive to corticosteroids. Nat Clin Pract Gastroenterol Hepatol. 2008(12):707-12. https://doi.org/10.1038/NCPGASTHEP1296 [ Links ]
30. Nakazawa T, Ando T, Hayashi K, Naitoh I, Ohara H, Joh T. Diagnostic procedures for IgG4-related sclerosing cholangitis. J Hepatobiliary Pancreat Sci. 2011;18(2):127-36. https://doi.org/10.1007/S00534-010-0320-2 [ Links ]
31. Mendes FD, Jorgensen R, Keach J, Katzmann JA, Smyrk T, Donlinger J, et al. Elevated serum IgG4 concentration in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2006;101(9):2070-5. https://doi.org/10.1111/J.1572-0241.2006.00772.X [ Links ]
32. Itoi T, Kamisawa T, Igarashi Y, Kawakami H, Yasuda I, Itokawa F, et al. The role of peroral video cholangioscopy in patients with IgG4-related sclerosing cholangitis. Journal of Gastroenterology. 2012;48(4):504-14. https://doi.org/10.1007/S00535-012-0652-6 [ Links ]
33. Saeki T, Ito T, Yamazaki H, Imai N, Nishi S. Hypocomplementemia of unknown etiology: an opportunity to find cases of IgG4-positive multi-organ lymphoproliferative syndrome. Rheumatology International. 2009;30(1):99-103. https://doi.org/10.1007/S00296-009-0925-4 [ Links ]
34. Saeki T, Kawano M, Mizushima I, Yamamoto M, Wada Y, Nakashima H, et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int. 2013;84(4):826-33. https://doi.org/10.1038/KI.2013.191 [ Links ]
35. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2015;74(1):14-8. https://doi.org/10.1136/ANNRHEUMDIS-2013-204907 [ Links ]
36. Zhang W, Stone JH. Management of IgG4-related disease. Lancet Rheumatol. 2019;1(1):e55-65. https://doi.org/10.1016/S2665-9913(19)30017-7 [ Links ]
37. Lanzillotta M, Fernàndez-Codina A, Culver E, Ebbo M, Martinez-Valle F, Schleinitz N, et al. Emerging therapy options for IgG4-related disease. Emerging therapy options for IgG4-related disease. Expert Rev Clin Immunol. 2021;17(5):471-83. https://doi.org/10.1080/1744666X.2021.1902310 [ Links ]
38. Okazaki K, Chari ST, Frulloni L, Lerch MM, Kamisawa T, Kawa S, et al. International consensus for the treatment of autoimmune pancreatitis. Pancreatology. 2017;17(1):1-6. https://doi.org/10.1016/J.PAN.2016.12.003 [ Links ]
39. Hart PA, Kamisawa T, Brugge WR, Chung JB, Culver EL, Czakó L, et al. Original article: Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut. 2013;62(12):1771. https://doi.org/10.1136/GUTJNL-2012-303617 [ Links ]
40. Omar D, Chen Y, Cong Y, Dong L. Glucocorticoids and steroid sparing medications monotherapies or in combination for IgG4-RD: a systematic review and network meta-analysis. Rheumatology (Oxford). 2020;59(4):718-26. https://doi.org/10.1093/RHEUMATOLOGY/KEZ380 [ Links ]
41. Deshpande V, Zen Y, Chan JKC, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Modern Pathology. 2012;25(9):1181-92. https://doi.org/10.1038/modpathol.2012.72 [ Links ]
Citación: Vargas-Marcacuzco HT, Junes-Pérez SI, Santos-Julián RE, Rocha-Bolaños RA, Matheus-Sairitupac J. Enfermedad relacionada con inmunoglobulina G4, un diagnóstico a tener en cuenta: a propósito de un caso. Revista. colomb. Gastroenterol. 2023;38(4):521-528. https://doi.org/10.22516/25007440.978
Recibido: 28 de Septiembre de 2022; Aprobado: 02 de Febrero de 2023
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
