










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkAgronomía Colombiana
Print version ISSN 0120-9965
Agron. colomb. vol.31 no.2 Bogotá May/Aug. 2013
PLANT BREEDING, GENETIC RESOURCES & MOLECULAR BIOLOGY
Genetic structure and evidence of putative Darwinian diversifying selection in the Potato yellow vein virus (PYVV )
Estructura genética y evidencia de una posible selección darwiniana diversificadora en el virus del amarillamiento de las venas de la papa (PYVV)
Giovanni Chaves-Bedoya1 Mónica Guzmán-Barney2 and Luz Yineth Ortíz-Rojas3
1Plantae Research Group, Department of Biology, Faculty of Basic Sciences, Universidad Francisco de Paula Santander. Cucuta (Colombia). gchavesbe@gmail.com2Plant Virus Laboratory, Institute of Biotechnology, Universidad Nacional de Colombia. Bogota (Colombia).
3Department of Basic Sciences, Universidad de los Llanos. Villavicencio (Colombia).
Received for publication: 15 Abril, 2013. Accepted for publication: 5 June, 2013.
ABSTRACT
The population structure and genetic variation of Potato yellow vein virus (PYVV) were estimated by analysis of the nucleotide and deduced amino acid sequence of the coat protein of 69 isolates, reported in GenBank, from Solanum tuberosum (ST ) and Solanum phureja (SP) hosts from different regions; predominantly Cundinamarca, Antioquia and Nariño, located in central and southwestern Colombia. Bioinformatics analysis revealed that despite the wide geographic distribution of different hosts and different collecting years, PYVV maintains a genetic similarity between 97.1 to 100.0%, indicating high spatial and temporal genetic stability of the major coat protein. No recombination events were found, but evidence was seen for the first time that this protein could be undergoing Darwinian diversifying selection.
Key words: Solanum tuberosum, viral proteins, genetic variability, Crinivirus.
RESUMEN
En este trabajo se estimó la estructura poblacional y variación genética del virus del amarillamiento de las venas de la papa (PYVV) infectando cultivos en Colombia por medio del análisis de 69 secuencias nucleotídicas y aminoácidos deducidos de la proteína mayor de la cápside (CP) reportados en el banco de genes (GenBank). Los aislamientos de PYVV fueron obtenidos de los hospederos Solanum tuberosum (ST ) y Solanum phureja (SP) en diferentes regiones de Colombia, predominantemente los departamentos de Cundinamarca, Antioquia y Nariño localizados en la región Central y Sur Oeste del país respectivamente. El análisis bioinformático reveló que a pesar de la amplia distribución geográfica de los hospederos y diferentes años de colecta, PYVV mantiene un similitud genética entre 97,1 y 100,0% indicando una gran estabilidad genética espacial y temporal en la CP. En este estudio no se detectaron eventos de recombinación, pero se presenta evidencia por primera vez de que esta proteína podría estar bajo selección darwiniana diversificadora.
Palabras clave: Solanum tuberosum, proteinas virales, variabilidad genética, Crinivirus.
Introduction
Worldwide, the potato is considered the fourth most important crop, after rice, wheat and maize. The potato can be infected by different viruses, including the Potato yellow vein virus (PYVV) which reduces the yield and quality of tubers (Salazar et al., 2000). PYVV has affected potato crops in Colombia for more than 50 years and has spread to neighboring countries in the Andean region. PYVV is classified as a tentative species of the Crinivirus genus in the Closteroviridae family. PYVV has a tripartite single strand and positive sense RNA genome. Virions are flexible, located in the phloem of the plant. The genome sequence indicates that the CP protein of PYVV consists of 756 nucleotides (252 aa) (Salazar et al., 2000). The virus is semi-persistent, transmitted by the greenhouse whitefly (Trialeurodes vaporariorum, Westwood) vector (Livieratos et al., 2004). PYVV is the causal agent of the Potato yellow vein disease (PYVD) which reduces the production in number of tubers by over 50% in the Solanum tuberosum group Andigena (Livieratos et al., 2004).
A characteristic of RNA viruses is that they have high genetic variability, due to the ability to generate large populations and the lack of a proof reading activity of RNA polymerase (Domingo and Holland, 1997). However, there are other factors that influence genetic diversity, such as genetic recombination, genomic rearrangement, genetic drift and natural selection (Garcia-Arenal et al., 2001). Genetic diversity among different viruses varies according to factors such as the virus-vector relationship, host range or geographic incidence. Characterization studies of genetic variability are of practical interest for the control of viruses; and strategies based on monogenic resistance are influenced by genetic variation of the pathogen (Vives et al., 2002).
Sequence analyses show that, in most instances, the selection acting on virus genes is negative. The degree of selection can be determinated from the ratio between nucleotide diversities at non-synonymous and synonymous positions (dN/dS). This ratio indicates the amount of variation in the nucleotides that results in variation in the encoded protein. Virus encoded proteins are not less constrained than those of their eukaryotic hosts and vectors, which suggests that the need to establish functional interactions with host and vector encoded factors is constraining the variability of virus encoded proteins (Garcia-Arenal et al., 2003). In virus genes, negative and positive selection may be acting in particular domains of the viral proteins, and are evidenced by detailed analyses of the encoding sequence. Poitive selection acts with resistance-breaking isolates (Garcia-Arenal et al., 2003).
Sequence analysis, in silico, of nucleotide or amino acids allowsfor the determination of possible phylogenetic relationships and similarities or differences among viral isolates, as has been reported for various species of viruses that infect plants using public sequences reported in the GenBank (Ge et al., 2007; Marco and Aranda, 2005; Martin et al., 2006; Rangel et al., 2011). Since there is currently no genetic variability or Darwinian selection analysis of PYVV using the CP sequences reported from different potato producing regions in Colombia, the aim of this study was to identify genetic variation in PYVV isolates infecting the potato in producing regions and to determine the presence of positive selection as an evolutionary force in PYVV based on 69 nucleotide sequences reported from different regions of Colombia. The results obtained for a region of 586 nucleotides within the coat protein gene covering a region encoding 195 aminoacids indicates low genetic variability, confirming previous studies (Guzmán et al., 2006; Offei et al., 2004; Rodriguez et al., 2010). Nevertheless, for the first time, we present evidence of positive Darwinian selection in two amino acids of the CP, suggesting this virus could be looking for change or speciation strategies.
Materials and methods
Origin of sequences
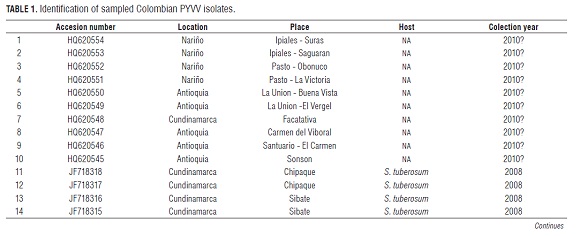
The nucleotide analysis was carried out with a total of 69 sequences of the coat protein (CP) of PYVV, which were obtained from the public database GenBank. The 68 sequences were obtained from major potato producing departments in Colombia; namely Antioquia, Boyaca, Cauca, Cundinamarca and Nariño. One sequence reported in Cajamarca, Peru (Livieratos et al., 2004) (GenBank AJ557129) was also used. Origin, year of collection and hosts are listed in Tab.1
Sequence edition for analysis
Since the nucleotide sequences of PYVV´s CP reported in GenBank have different lengths, for the analysis, all sequences were aligned using the ClustalW program implemented in the program MegAlign™ (DNAS tar, Madison, WI) package for sequence analysis, version 7.2.2 and were adjusted to a length of 586 nucleotides flanked by the highly conserved amino acid sequences KDDSYNLDL and DLTANYLFK . The CP sequences of PYVV starting with GenBank code HQ (i.e., HQ620554) were adjusted from nucleotide position 86 to 668. Sequences starting with code JF (i.e., JF718316) were adjusted from nucleotide position 109 to 696. Sequences starting with code GQ (i.e., GQ397987) were adjusted from nucleotide position 1 to 588 and sequences starting with code AJ (i.e., 586113) were adjusted from nucleotide position 76 to 661. We assumeda high qualityfor the sequences deposited in GenBank, which were generated by third parties.
Phylogenetic analysis
The phylogenetic relationship of the nucleotide sequences was inferred by the Neighbor-Joining method (Saitou and Nei, 1987). Evolutionary distances were calculated using the Kimura 2-parameter method (Kimura, 1980), using 1000 replications to estimate the confidence of the taxon grouping in tree branches (Felsenstein, 1985). All positions containing gaps and missing data were eliminated. The evolutionary analysis was performed in the program MEGA 5 (Tamura et al., 2011).
Recombination and selection
The search for putative recombination events was done using the genetic algorithm for recombination detection (GA RD) (Kosakovsky Pond et al., 2006). The nucleotidesubstitution model was selected automatically before being applied to the site-recombination analysis. The search for amino acids undergoing selection was performed using the algorithms FEL (fixed effects likelihood), REL (random effects likelihood), (Kosakovsky Pond and Frost, 2005) and MEME (mixed effects model of evolution) implemented in the HyPhy program (hypothesis testing using phylogenies) (Pond and Frost, 2005) in the datamonkey server (Delport et al., 2010). This method allows for the identification of codons undergoing positive selection and removes the assumptions about the demographics associated with other statistical selection tests (Cavatorta et al., 2008).
Results and discussion
Nucleotide similarity
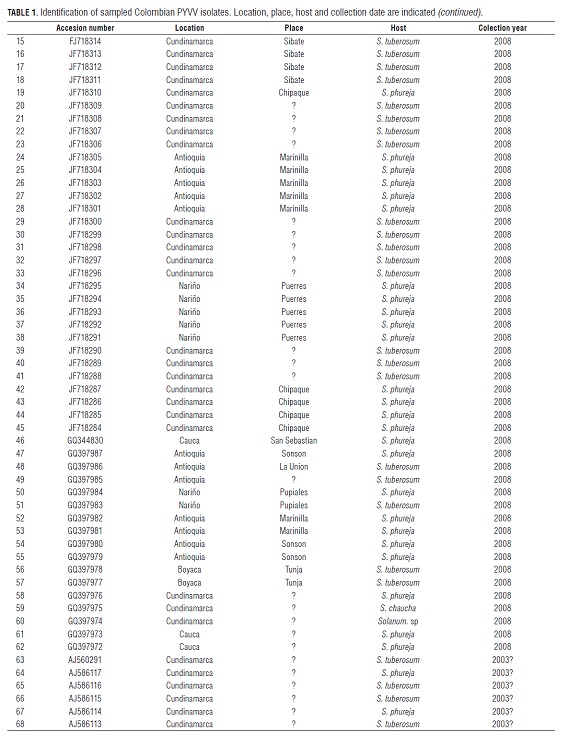
Unlike previous reports (Guzmán et al., 2006; Offei et al., 2004), this is the first study that analyzed the genetic variability of the CP of PYVV using 68 sequences from different geographical regions of Colombia, hosts and different years of sampling. All sequences are available in GenBank (Tab.1). Nucleotide similarity among the CP ranged from 97.1 to 100.0%, with 97.3% being the most frequent value, as indicated by the graph of frequency obtained from 2,278 nucleotide paired comparisons (data not shown). The results indicate that Colombian PYVV isolates exhibit high genetic stability over time and among different departments and years. Several collected PYVV isolates, either in different years or departments, had 100% nucleotide similarity (Tab.2).
In an attempt to discriminate potential PYVV genetic groups circulating in Colombia, we built a phylogenetic tree using 32 PYVV haplotypes that were deducted in the program SNAP (Price and Carbone, 2005) . The phylogenetic tree of PYVV haplotypes (Fig.1) does not show evidence of genetic groups, indicating that PYVV in Colombia is very homogeneous without clear genetic groups according to geographical precedence, host or year. More Colombian PYVV sequences are needed in order to better estimate phylogenetic inferences.

Haplotypes and related isolates are listed in Tab.3. The most common PYVV haplotype in Colombia is identified as H28, which includes isolates obtained from different potato species sampled in the departments of Cundinamarca, Antioquia and Cauca. Geographically, the departments of Antioquia and Cundinamarca are adjacent to one another in the central region of Colombia and the Cauca department is located in the southwest of the country (Fig.1).

High nucleotide similarity values between PYVV isolates suggest: first, a high spatial and temporal genetic stability and second, the possible movement between departments of tubers infected with the virus. Indeed, the presence of identical PYVV haplotypes infecting potato crops in different departments of Colombia indicates that there is an urgent need to improve the quality of potato tubers with a certified seed production program. It is also important to regulate and limit the transport of seeds between departments and borders, because PYVV is a virus that can be transmitted through tubers (Salazar et al., 2000). The transfer of potato tubers between departments is a practice that is often used among Colombian farmers, but the detection of the virus by visual analysis is not possible because the infected tubers have no apparent symptoms, making it difficult to predict whether or not they have the virus.
Control of viral spread remains the most efficient method to reduce viral diseases in potato seed production Within the family Closteroviridae, other viruses have been reported with low CP genetic diversity from geographically distant isolates (Alicai et al., 1999; Rubio et al., 2001a; Rubio et al., 2001b; Rubio et al., 1999). Several references pointing to similar results indicate that low heterogeneity is the norm for viruses in the genus Crinivirus.
Selection analysis
Nucleotide or amino acid selection can be exercised to maintain the primary, secondary or tertiary structural characteristics in the viral genome that are important for replication, such as the 3´non coding genomic regions of the single strand RNA virus. Another group of selection factors is associated with the host plant. The differentiation of natural populations according to the host plant can also be taken as evidence of host-associated selection (Garcia- Arenal et al., 2001). Most positive-strand plant RNA viruses are adaptedtoinfection of plant hosts. The comparison of genetic maps of representative viruses has revealed genes in plant viral genomes that appear to be essential adaptations needed for success fulinvasion and dispersal throughout their plant hosts. (Goldbach et al., 1994).
A non-synonymous substitution rate (dN) significantly higher than the percentage of synonymous substitution (dS), or w = dN/dS greater than 1, points to positive Darwinian selection (Delport et al., 2009). Since the selection acting on viral genes is negative in most cases, (Garcia-Arenal et al., 2003), and positive selection is less frequent (Gojobori et al., 1990), we focused on positively selected codons.
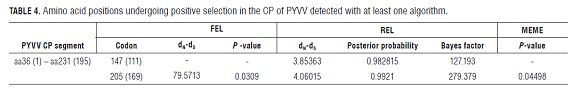
The prior selection analysis was searched for possible recombination events, because it may contribute to a false inference of positive selection (Scheffler et al., 2006). With no evidence of recombination, the nucleotide segment was not subdivided for further selection analysis (Scheffler et al., 2006). For the CP of PYVV, three different algorithms, designed to detect selection in a particular codon, coincided in suggesting that codon 205 is undergoing diversifying selection (Fig.2).
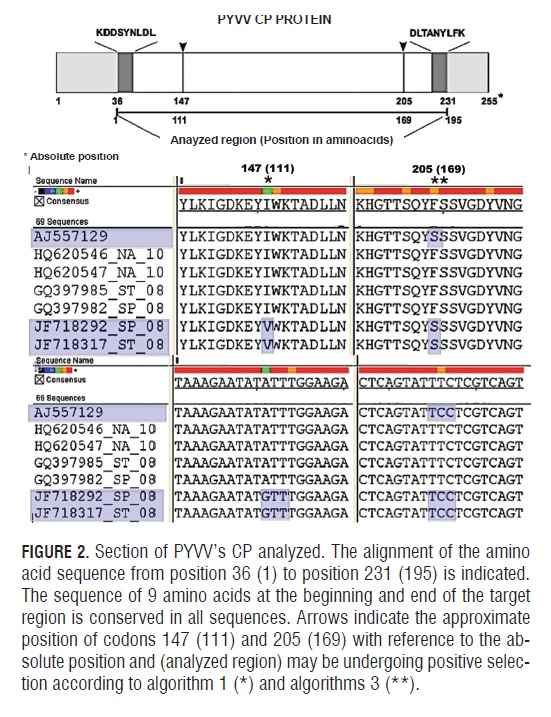
Positive selection in codon 147 is supported only by the REL algorithm. The amino acid at position 205 in the majority of the isolates corresponded to a phenylalanine, except for isolates JF718317 (ST ) from Cundinamarca, JF718292 (SP) from Nariño and AJ557129 (ST ) from Peru, which codified for a serine.
The dN/dS ratio of CP sequences analyzed in the MEGA program, where dN is the ratio of non-synonymous substitutions and dS is the ratio of synonymous substitutions, is 0.19. This value is in the range for viruses that infect plants (Garcia-Arenal et al., 2001). dN/dS = 0.19 indicates a negative selection pressure for the amino acid change in this genomic region. However, the results of selection pressure which fall in the average dN/dS range of a region of interest may have a poor statistical power to detect positive selection because only a few sites may be undergoing selection (Kosakovsky Pond and Frost, 2005). In a site to site search for selection, algorithms FEL (P-value 0.030), REL (Bayes Factor 279 379) and MEME (P-value 0.044) indicated that there is enough statistical evidence to suggest that codon 169 of the alignment is undergoing positive selection (Fig.2). Unlike FEL and MEME, REL analysis further suggested that codon 111 may also be undergoing positive selection (Tab.4).
Isolates that have amino acid changes at two positions undergoing putative diversifying selection included JF718292 (SP), JF718317 (ST ) and AJ557129 (ST ), the latter reported in Peru. For Colombian PYVV isolates, at position 147 (111), there was a change of isoleucine to valine due to the transition of GTT ? ATT . However, in this position, 24 other isolates presented a valine. For position 205 (169), only isolates JF718292, JF718 317 and AJ557129 had the amino acid change of phenylalanine to a serine, due to the transition of TT C → TCC.
Phenylalanine has been reported as an important amino acid in the differentiation of viral strains in Citrus tristeza virus (CTV) at position 124 of the CP. This aminoacid causes the epitope responsible for the reaction with antibody MCA 13, which differentiates between severe and soft strains of CTV with 95% confidence (Pappu et al., 1993; Permar et al., 1990). Our in silico results could be coincidental, and the amino acid variation of serine or phenylalanine could indicate a change in epitope recognition for PYVV. However, this is a hypothesis that should be demonstrated because in this virus, there are no reports on strains expressing different symptoms. Amino acids identified as undergoing positive selection in PYVV could be in a region of interaction with proteins of the vector and positive selection reflects gain or loss of affinity for the interaction with the vector, since no correlation between the encoded amino acid and the host was found.
Conclusions
The low heterogeneity found in PYVV is possibly due to its rapid expansion as a result of whitefly population growth and expansion, or similar selection pressures in the different species of Solanum.
The proposed functions of the positively selected aminoacids in the CP of PYVV are speculative. We are extrapolating the function of another two suggested amino acids undergoing positive selection in an unrelated gene of virus species belonging to a different genus in the family Closteroviridae. Not much can be concluded from the small number of amino acid variants without a biological relationship. Although this does suggest selection pressure, one cannot guess what the selection would be without an obvious biological trait. For now, this change only suggests some limited variation among PYVV isolates. Further studies must be carried out to determinate the biological significance of this variation.
Literature cited
Alicai, T., N.S. Fenby, R.W. Gibson, E. Adipala, H.J. Vetten, G.D. Foster, and S.E. Seal. 1999. Occurrence of two serotypes of sweet potato chlorotic stunt virus in East Africa and their associated dierences in coat protein and HSP70 homologue gene sequences. Plant Pathol. 48, 718-726. [ Links ]
Cavatorta, J.R., A.E. Savage, I. Yeam, S.M. Gray, and M.M. Jahn. 2008. Positive Darwinian selection at single amino acid sites conferring plant virus resistance. J. Mol. Evol. 67, 551-559. [ Links ]
Delport, W., A.F. Poon, S.D. Frost, and S.L. Kosakovsky Pond. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455-2457. [ Links ]
Delport, W., K. Scheffler, and C. Seoighe. 2009. Models of coding sequence evolution. Brief. Bioinform. 10, 97-109. [ Links ]
Domingo, E. and J.J. Holland. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151-178. [ Links ]
Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783-791. [ Links ]
Garcia-Arenal, F., A. Fraile, and J.M. Malpica. 2001. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 39, 157-186. [ Links ]
Garcia-Arenal, F., A. Fraile, and J.M. Malpica. 2003. Variation and evolution of plant virus populations. Int. Microbiol. 6, 225-232. [ Links ]
Ge, L., J. Zhang, X. Zhou, and H. Li. 2007. Genetic structure and population variability of tomato yellow leaf curl China virus. J. Virol. 81, 5902-5907. [ Links ]
Gojobori, T., E.N. Moriyama, and M. Kimura. 1990. Molecular clock of viral evolution, and the neutral theory. Proc. Natl. Acad. Sci. USA 87, 10015-10018. [ Links ]
Goldbach, R., J. Wellink, J.Verver, A. Van Kammen, D. Kasteel, and J. Van Lent. 1994. Adaptation of positive-strand RNA viruses to plants. Arch. Virol. Suppl. 9, 87-97. [ Links ]
Guzmán, M., E. Ruiz, N. Arciniegas, and R.H. Coutts. 2006. Occurrence and variability of Potato yellow vein virus in three departments of Colombia. J.Phytopatol. 154, 748-750. [ Links ]
Kimura, M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111-120. [ Links ]
Kosakovsky Pond, S.L. and S.D. Frost. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208-1222. [ Links ]
Kosakovsky Pond, S.L., D. Posada, M.B. Gravenor, C.H. Woelk, and S.D. Frost. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891-1901. [ Links ]
Livieratos, I.C., E. Eliasco, G. Muller, R.C. Olsthoorn, L.F. Salazar, C.W. Pleij, and R.H. Coutts. 2004. Analysis of the RNA of Potato yellow vein virus: evidence for a tripartite genome and conserved 3'-terminal structures among members of the genus Crinivirus. J. Gen. Virol. 85, 2065-2075. [ Links ]
Marco, C.F. and M.A. Aranda. 2005. Genetic diversity of a natural population of Cucurbit yellow stunting disorder virus. J. Gen. Virol. 86, 815-822. [ Links ]
Martin, S., M.L. Garcia, A. Troisi, L. Rubio, G. Legarreta, O. Grau, D. Alioto, P. Moreno, and J. Guerri. 2006. Genetic variation of populations of Citrus psorosis virus. J. Gen. Virol. 87, 3097-3102. [ Links ]
Offei, S.K., N. Arciniegas, G. Muller, M. Guzman, L.F. Salazar, and R.H. Coutts. 2004. Molecular variation of Potato yellow vein virus isolates. Arch. Virol. 149, 821-827. [ Links ]
Pappu, H., S. Pappu, C. Niblett, R. Lee, and E. Civerolo. 1993. Comparative sequence analysis of the coat proteins of biologically distinct citrus tristeza closterovirus isolates. Virus Genes 7, 255-264. [ Links ]
Permar, T.A., S.M. Gransey, D.J. Gumpf, and R.F. Lee. 1990. A monoclonal antibody wich discriminates strains of Citrus tristeza virus. Phytopathology 80, 224-228. [ Links ]
Pond, S.L., and S.D. Frost. 2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531-2533. [ Links ]
Price, E.W. and I. Carbone. 2005. SNA P: workbench management tool for evolutionary population genetic analysis. Bioinformatics 21, 402-404. [ Links ]
Rangel, E.A., A. Alfaro-Fernandez, M.I. Font-San-Ambrosio, M. Luis-Arteaga, and L. Rubio. 2011. Genetic variability and evolutionary analyses of the coat protein gene of Tomato mosaic virus. Virus Genes 43, 435-438. [ Links ]
Rodriguez, P., G. Chaves-Bedoya, L. Franco-Lara, and M. Guzmán. 2010. Low molecular variability of Potato yellow vein virus (PYVV) isolates of Solanum phureja and Solanum tuberosum from Colombia. Phytopathology 100, s176. [ Links ]
Rubio, L., Y. Abou-Jawdah, H.X. Lin, and B.W. Falk. 2001a. Geographically distant isolates of the crinivirus Cucurbit yellow stunting disorder virus show very low genetic diversity in the coat protein gene. J. Gen. Virol. 82, 929-933. [ Links ]
Rubio, L., M.A. Ayllon, P. Kong, A. Fernández, M. Polek, J. Guerri, P. Moreno, and B.W. Falk. 2001b. Genetic variation of Citrus tristeza virus isolates from California and Spain: evidence for mixed infections and recombination. J. Virol. 75, 8054-8062. [ Links ]
Rubio, L., J. Soong, J. Kao, and B.W. Falk. 1999. Geographic distribution and molecular variation of isolates of three whitefly-borne closteroviruses of cucurbits: lettuce infectious yellows virus, cucurbit yellow stunting disorder virus, and beet pseudoyellows virus. Phytopathology 89, 707-711. [ Links ]
Saitou, N. and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406-425. [ Links ]
Salazar, L.F., G. Miller, M. Querci, J.L. Zapata, and R.A. Owens. 2000. Potato yellow vein virus: its host range, distribution in South America and identification as a crinivirus transmitted by Trialeurodes vaporariorum. Ann. Appl. Biol. 137, 7-19. [ Links ]
Scheffler, K., D.P. Martin, and C. Seoighe. 2006. Robust inference of positive selection from recombining coding sequences. Bioinformatics 22, 2493-2499. [ Links ]
Tamura, K.,D. Peterson, N. Peterson, G. Stecher, M. Nei, and S. Kumar. 2011. MEGA 5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28(10), 2731-9. [ Links ]
Vives, M.C., L. Rubio, L. Galipienso, L. Navarro, P. Moreno, and J. Guerri. 2002. Low genetic variation between isolates of Citrus leaf blotch virus from different host species and of different geographical origins. J. Gen. Virol. 83, 2587-2591. [ Links ]
