










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.24 no.3 Bicaramanga Sept./Dec. 2011
Helicobacter pylori: revisión de los aspectos
fisiológicos y patológicos
Jorge Luís Suárez Guerrero*
Genny Carolina Reyes Vera **
Lina del Mar Herreros Rosas ***
*Estudiante VI nivel. Escuela de Medicina. Integrante Grupo de Investigación en Genética Clínica. Miembro SEIMED-UIS. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
**Estudiante VI nivel. Escuela de Medicina. Miembro SEIMED-UIS. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
***Estudiante IV nivel. Escuela de Medicina. Integrante Grupo de Investigación en Genética Clínica. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
Correspondencia: Sr. Suárez Calle 4 # 12 - 56 Nuevo Villabel. Bucaramanga. Colombia. e-mail: Jorgesuarezg_@hotmail.com, jorgesuarezg@gmail.com.
Artículo recibido el 20 de Septiembre de 2011 y aceptado para publicación el 3 de Diciembre de 2011.
RESUMEN
Helicobacter pylori es una bacteria con forma helicoidal, que vive únicamente en el estómago humano. Aunque la relación del bacilo con el epitelio es de antaño debido a que gran parte de la población se halla colonizada, tan solo cerca del 10% de las personas infectadas desarrollan ciertas patologías, entre ellas la úlcera duodenal, el adenocarcinoma estomacal y el linfoma tipo MALT. En las últimas décadas, la investigación sobre el Helicobacter pylori se ha incrementado, y poco a poco, se ha deslumbrando más sobre la fisiopatología de este microorganismo, como la presencia de genes involucrados en la patogenicidad vacA, cagA babA y sabA, indicando con ello que no es precisamente una bacteria totalmente inocua, pero tampoco el del mayor patógeno en el epitelio estomacal. Mediante la presente revisión se pretende abarcar algunos aspectos sobre la bacteria, su relación y beneficios con el huésped, así como los procesos patológicos que puede desencadenar su accionar. (MÉD.UIS. 2011;24(3):275-82).
Palabras Clave: Helicobacter pylori. Úlcera péptica. Linfoma tipo MALT. Linfoma de Células B de la Zona Marginal.
ABSTRACT
Helicobacter pylori: review of physiologic and patologic aspects.
Helicobacter pylori is a spiral-shaped bacterium, that lives only in the human stomach. Although the relationship of the bacillus in the epithelium has been since ancient because most of the population is colonized, only about 10% of infected people develop certain diseases, including duodenal ulcer, stomach adenocarcinoma and lymphoma MALT. In recent decades, research on Helicobacter pylori has increased, and little by little, has dazzled more about the pathophysiology of this organism, as the presence of genes involved in pathogenicity vacA, cagA, babA and sabA thereby indicating that is just a completely harmless bacteria, but neither of the major pathogen in stomach epithelium. Through this review is to cover some aspects of the bacteria, their relationship and benefits to the host and the pathological processes that can trigger their actions. (MÉD.UIS. 2011;24(3):275-82).
Keywords: Helicobacter pylori. Peptic ulcer. Lymphoma MALT. Lymphoma. B-Cell. Marginal Zone.
INTRODUCCIÓN
El Helicobacter pylori (H. pylori) al microscopio óptico y con el uso de colorantes tales como hematoxilina-eosina, tinción de Warthin-Starry o el Giemsa modificada1, se observa en medios orgánicos como un bacilo gram negativo con forma curvilínea y multiflagelado cuyo tamaño varía a lo ancho entre 0,5 a 1 µm y a lo largo desde 2,5 hasta 6,5 µm, omitiendo el tamaño de los flagelos que pueden ser de hasta 30 µm1-3. El estudio mediante pruebas enzimáticas ha permitido identificar al H. pylori y catalogarlo como catalasa, oxidasa y ureasa positivo4.
En el ser humano esta colonización abarca cerca del 50% de la población mundial, especialmente los habitantes en los países en vía de desarrollo5-9. Esta inicia desde la infancia10,11 indicando con ello un equilibrio entre la bacteria, su ambiente así como la respuesta inmunológica, con ello revelando un proceso coevolutivo7,10.
El estómago era considerado un sitio libre de bacterias, ya que por su ambiente ácido era improbable que alguna creciera allí , sin embargo esta idea cambió desde el descubrimiento del microorganismo por los doctores Robin Warren y Barry Marshall en 198312, y desde entonces prácticamente se ha reescrito la historia de la gastroenterología en cuanto a patologías en este ámbito se refiere.
Sin duda, uno de los rasgos más notable del H. pylori es la gran diversidad genética que han descubierto los diferentes estudios hasta la fecha5,13-7, incluyéndose la diversidad de la secuencia conservadas de genes que puede llegar hasta un 6%, la variabilidad de los elementos móviles de ADN, y el hecho que en el ser humano se han registrado dos cepas, clasificadas como tipo I y tipo II1,10,16,17. Gracias a esta diversidad genética, se ha favorecido el interés por seguir las investigaciones, no solo desde el punto de vista genético, sino también desde la epidemiología, clínica, y salud pública entre otros. Como consecuencia, se ha generado una serie de controversias entre especialistas de todas partes del mundo, sustentando ideas opuestas e incluso radicales, que van desde desconocer tal agente como el responsable directo de diversas patologías hasta señalarle como único factor causal o de riesgo1.
El objetivo de este artículo es realizar una revisión de los potenciales beneficios fisiológicos, así como los riesgos causados por el microorganismo.
EL HELICOBACTER PYLORI Y EL EPITELIO GÁSTRICO
Vías de Transmisión: la transmisión del H. pylori es principalmente por vías oral-oral y fecal-oral, la primera debido a la presencia transitoria de bacterias en la boca que se pueden trasmitir por compartir utensilios y la segunda, por beber fuentes de agua o alimentos contaminados los cuales actúan como reservorios temporales para la bacteria. Esta capacidad de transmisión se debe en primera instancia a la capacidad de supervivencia temporal al medio ambiente que presenta la bacteria20 y por la presencia de factores de riesgos que facilitan la propagación del microorganismo, tales como: las altas tasas de hacinamiento, malas condiciones socioeconómicas y deficientes condiciones de higiene. La infección se da principalmente durante la infancia11,21,22, aunque las manifestaciones se presentan en la vida adulta tardía23.
Hábitat: hasta el momento, se sabe que el epitelio gástrico humano es el único nicho ecológico del microorganismo, por ello se ha estimado que la bacteria no es exclusivamente un patógeno, sino más bien un agente anfibiótico, es decir, puede vivir en mutualismo con algunos huéspedes, mientras que en otros es capaz de desarrollar enfermedad bajo ciertas condiciones1,2, como con los productos de los genes vacA, cagA babA y sabA6,24-33.
En el estómago humano, la región anatómica denominada antro es donde se lleva a cabo la mayor colonización por parte del H. pylori3. Debido a la presencia de los flagelos en sus polos, puede desplazarse hasta entrar en contacto con el epitelio gástrico, al que se puede unírsele gracias a la presencia de adhesinas como la hialuronidasa que le permite interactuar con receptores epiteliales como los TLR (Toll Like Receptors)7, así como al glucocáliz ubicado cerca de la zónula ocludens del epitelio gástrico; de esta forma no solo asegura su unión, sino que evita ser desplazado a zonas del tracto digestivo en donde no puede colonizar1,3,34,35. Todo este proceso de colonización puede tardar hasta una semana, ya que entre otras cosas debe activar la ureasa para convertir la urea en amoniaco para evitar la acción del ácido clorhídrico, así como escapar a la respuesta inmune1,36,37.
Muchas veces este éxito en la unión no solo despierta una respuesta inflamatoria leve o "benigna" y el desarrollo de cambios en el ambiente estomacal que protege al organismo de otros patógenos. También favorece la aparición de diferentes patologías, iniciando con lesiones leves del epitelio como disminución en la producción de moco o atrofia de las microvellosidades, hasta el desarrollo del adenocarcinoma gástrico. Estas últimas ocurren, especialmente ante la presencia de cepas con islas de patogenicidad, así como ante personas que desarrollan una respuesta inmunitaria inadecuada7,25.
GENES ASOCIADOS A PATOGENICIDAD
La capacidad de patogenia gastroduodenal por parte del H. pylori se debe en gran parte a su capacidad de sintetizar los productos procedentes de los genes vacA, cagA, babA y sabA5,6,10,28,32,38-40. En cuanto al gen vacA (Citotoxina Activa Vacoulizante), se sabe, que su presencia induce la formación de vacuolas, aunque no todas las cepas con este gen desarrollan estos cambios epiteliales. Por su parte el gen cagA (Citotoxina Asociada al gen A) induce la expresión de IL-8 en las células epiteliales1.El gen babA (Adhesina de Unión al Antígeno del Grupo Sanguíneo) sintetiza una proteína que permite la unión del microorganismo al antígeno de Lewis1,32. El gen sabA (Adhesina de Unión al Ácido Siálico) tiene función similar al babA al sintetizar una proteína de unión, pero esta lo hace con el antígeno Sialico de Lewis29. También se ha indicado un posible daño del ADN celular, como consecuencia de la acción de los productos del gen vacA, y del cagA, debido a la alteración de los mecanismos de reparación del ADN del hospedero41. En resumen la presencia de ambos genes y sus productos son factores de riesgo importantes para el desarrollo de patologías gastrointestinales severas.
Gen vacA: el gen vacA, genera una proteína que induce la formación de vacoulas en el epitelio del hospedero. Esta proteína presenta variantes en la región de señalización del péptido (s1, s2), así como en la región media (m1, m2) y dependiendo de la combinación de ellos se van a producir efectos tóxicos en mayor o menos medida, así por ejemplo el genotipo con s1/m1 produce más toxina vacuolizante que las cepas con el genotipo s2/m2, explicando hasta cierto punto porque en promedio solo el 50% de las cepas vacA positivas desarrollan vacuolas en el epitelio6,28,38,40,42,43.
Gen CagA: el gen cagA se ubica dentro de la isla de patogenicidad cag. La función del gen es modulda mediante fosforilación de receptores tirosin-cinasa, exactamente en las proteínas src, que a su vez afecta la cascada de señalización de la de la PI3K, PLC, NF- kB y Ras18,44,45. En otras palabras, codifica la proteína que altera las vías de señalización intracelular de los linfocitos B, facilitando así la sobreexpresión de genes antiapoptosis Bcl-2 y Bcl-x, favoreciendo la aparición de linfomas tipo MALT41,45-7. Además de ello, favorece la producción de IL-8, que estimula la respuesta inflamatoria en el epitelio gástrico. Algunos estudio asocian la posible presencia del gen cagA con el desarrollo de cáncer de las vías biliares, debido la acción de las células inflamatorias, así como la alteración de la proliferación y la apoptosis de las células biliares44.
Gen babA: las cepas de H. pylori, pueden tener dos grupos del alelos del gen bab, uno denominado babA y el otro. Solo la proteína de membrana sintetizada por el gen babA, tienen la capacidad de unirse al antígeno de Lewis B, permitiendo la colonización de la mucosa gástrica indemne, al facilitar la unión de la bacteria con el epitelio del hospedero31,32. Además se ha relacionado babA, con la presencia de los genes vacA y cagA,32,33.
Gen sabA: el gen produce una proteína de membrana que le permite al H. pylori unirse al el antígeno Siálico de Lewis, el cual está presente en grandes cantidades cuando se desencadena una respuesta inflamatoria29,30. El gen sabA y su producto se han encontrado en cepas relacionadas con el desarrollo de metaplasia intestinal, atrofia gástrica y adenocarcinoma29.
EFECTOS FISIOLÓGICOS EN EL ORGANISMO HUMANO
Dentro de los supuestos efectos benéficos de la infección por HP, se ha destacado la producción de amonio a partir de urea, la cual sirve como tampón para aumentar el pH estomacal, especialmente sobre la unión gastroesofágica. Esta acción de aumento del pH, también se ve favorecida, por la modificación en la producción de gastrina48, principalmente porque el H pylori secreta N-alfa- metilhistamina, y estimula la producción de histamina, ambos agonistas de receptores de histamina, y cuyo resultado final es una respuesta de retroalimentación negativa en la producción de ácido clorhídrico23. En otras palabras, el cambio del pH estomacal puede prevenir el daño a la mucosa del esófago y con ello evita la aparición de lesiones como el esófago de Barret, así como el posible desarrollo de un adenocarcinoma gástrico23,49-51. Se debe aclarar que hay estudios que no relacionan estos beneficios con la presencia o ausencia del H. Pylori52, mientras que otros establecen ciertas condiciones para obtener estos cambios como sería la ubicación geográfica del microorganismo en el estómago, la ausencia de úlcera duodenal, entre otros23,53,54. Por ello este tema sigue siendo algo controvertido y debatible.
Otros de los beneficios es que induce la producción de ácido clorhídrico en otras regiones del estómago, generando un ambiente hostil para otras bacterias, evitando que agentes mucho más patógenos como la Salmonella typhi, infecten el epitelio estomacal ya sea por acceso oral o cólico1,49,50. Otro nuevo aspecto es, la tolerancia a este microorganismo, y su función disminuye los requerimientos del sistema inmune para mantener la homeostasis estomacal, ya que el H. pylori favorece procesos inflamativos leves, indicando con ello una activación permanente del sistema inmunológico1.
EFECTOS PATOLÓGICOS EN EL ORGANISMO HUMANO
Si bien, se considera el H. pylori trae beneficios para el huésped, también se ha relacionad su presencia con el desarrollo de patologías tales como la dispepsia, la gastritis crónica, la úlcera péptica, y el cáncer de estómago38,40,55-7. La bacteria, puede actuar como patógeno, cuando daña directamente el epitelio gástrico o cuando desarrolla procesos de inflamación crónica, que pueden complicarse, es decir hay un daño irreversible del epitelio gástrico. Si bien la infección está presente en más de la mitad de la población, la mayoría de las personas infectadas desarrollan gastritis asintomáticas más que patologías severas1. Sin embargo la gastritis asintomática coincide con daños más notorios que pueden favorecer el desarrollo de adenocarcinoma18,58 y de linfomas tipo MALT59. La erradicación exitosa de la bacteria, favorece la desaparición de los procesos inflamatorios, la regeneración y reparación del tejido afectado48.
Cambios del epitelio gástrico: una vez se inicia la colonización, se puede observar en cortes microscópicos los cambios estructurales que sufren las denominadas fositas o foveolas conformadas de epitelio cilíndrico. La bacteria evita el desarrollo de la respuesta inmune gracias a la presencia de lipopolisacaridos similares a los del epitelio, brindándole una especie de camuflaje por su bajo poder antigénico, haciendo referencia concreta al antígeno de Lewis; sin embargo este puede ocasionar respuesta inmune cruzada. También favorece el aumento de la secreción de grelina60 y la reducción en la secreción de leptina, lo cual podría suponer una alteración del apetito favoreciendo la obesidad, aunque la relación causa efecto no está bien dilucidada10.
Infección e inflamación: la producción de ácido gástrico se ve alterada en grados variables debido a las alteraciones en el equilibrio de la gastrina y somatostatina61. Esto se traduce en un aumento del pH hasta valores casi neutros sobre la superficie de los enterocitos, lo que favorece el desarrollo de la respuesta inflamatoria, así como la disminución de la secreción gástrica, que bajo ciertas condiciones es un factor de riesgo para el desarrollo de patologías61. Otro de los factores claves es la presencia de antígenos, como el de Lewis, que pese a permitir ocultarse de la respuesta inmune, puede favorecer una respuesta inmune cruzada, que atacaría no solo al microorganismo, sino a las células epiteliales1. Por otro lado, el daño a la mucosa también se puede dar de forma indirecta al combinarse el amonio liberado por la bacteria con el ion cloruro de los polimorfonucleares formando monocloramina, un compuesto citotóxico para el epitelio que de igual manera favorece la respuesta inflamatoria en el tejido.
En los primeros estadios, el daño estimula la presencia de polimorfonucleares neutrófilos que desencadenan la reacción inflamatoria aguda, la cual debería contener el daño generado por el microorganismo3. A su vez la interacción del microorganismo con el CD74 de las células epiteliales gástricas, induce la activación del NF-kB y con ello la producción de IL-818,62, así como la producción de IL-12 la cual favorece la respuesta inmune celular específica. Otra de las características es la presencia de linfocitos T, quienes son reclutados por la interacción entre el ligando CCR6 y la quimioquina CCL20, favoreciendo así la inflamación y la apoptosis celular63.
Estos procesos inflamatorios, favorecen la infiltración al epitelio estomacal acompañados de la liberación de citotoxinas, enzimas y radicales libres, causando un daño al ADN de las células de la mucosa, que conllevan hacia la apoptosis celular, ocasionando así daños generalizados en el epitelio gástrico. Otro de los factores que interviene en estos procesos inflamatorios es el Factor de Necrosis Tumoral alfa, FNT-α59. Además de estas interleucinas se ha relacionado la activación de genes proinflamatorios, como el COX-2 (Ciclooxigenasa-2), y el iNOS (Oxido nítrico Sintasa Inducible), los cuales se relacionan con la vía de señalización del Ras y el factor de activación AP-1, que involucra a sus vez la activación de c-fos y c-jun, dentro de las células epiteliales. La importancia de estos factores no es solo su estimulación por parte del epitelio cuando hay infección por el microorganismo, sino que además su activación se ha relacionado con el desarrollo de adenocarcinomas gástricos58.
Cuando el microorganismo no es debidamente erradicado o la respuesta aguda llega a ser tan severa que continua lesionando el epitelio, se desencadena un proceso inflamatorio crónico, que conlleva al desarrollo de las patologías relacionadas3. En cuanto a la respuesta inmune, se ha relacionado la ausencia del Complejo Mayor de Histocompatibilidad HLA-DQA1 con el desarrollo de atrofia gástrica, así como el desarrollo del adenocarcinoma tipo intestinal64, mientras que la presencia del HLA-DQB1 se relaciona con una mayor posibilidad de desarrollar adenocarcinoma gástrico de tipo intestinal65,66.
Gastritis crónica: es una enfermedad inflamatoria crónica de la mucosa estomacal, que puede generar desde la atrofia leve de la mucosa hasta el desarrollo de adenocarcinomas67,68. Las causas de esta atrofia son múltiples, sin embargo la mayoría son debidas a la presencia del H. pylori, pues la respuesta inflamatoria desarrollada induce la apoptosis del epitelio gástrico62,63. Otras causas de gastritis crónica son las de origen inmunológico como anticuerpos citotóxicos o contra el factor intrínseco, el consumo recurrente de alcohol, el tabaquismo, y la radiación67,68. Si bien la gastritis crónica se caracteriza por estar eritematosa con piegues aumentados de tamaño en un comienzo, estos suelen volverse aplanados y finos por el mencionado daño epitelial. Morfológicamente la gastritis crónica, tiene varios patrones de presentación sin embargo con la infección por la bacteria la tendencia es a encontrar la lesión limitada a la zona del antro67,68.
Úlcera péptica: en términos generales, la úlcera se considera una pérdida de la continuidad del epitelio. En el caso de la úlcera péptica, la ubicación más frecuente es la primera porción del duodeno. Esta lesión se ha asociado a diferentes patologías, pero la principal relación es con la presencia del bacilo bien sea en el antro o en el duodeno. La presencia el en antro favorece el aumento en la cantidad de ácido que llega al duodeno lo cual daña el epitelio48, mientras que la presencia en el duodeno, favorece la formación de úlceras mediante la acción de las células dendríticas y la subsiguiente activación de la respuesta inmune69. La severidad de la úlcera va a depender en gran medida de la presencia de cepas cagA positivas42 así como con la carga bacteriana, y la edad del paciente, presentando un curso clínico diferente en adultos mayores, caracterizado por epigastralgía así como complicaciones de las úlceras70. Esta es una de las patologías que se ve favorecida con la erradicación del microorganismo, entre otras cosas por la disminución de la respuesta inmune inflamatoria y el daño generado al epitelio gástrico.
Cáncer gástrico: se considera que las personas infectadas con H. Pylori presentan un riesgo seis veces mayor de desarrollar cáncer gástrico, debido a que el microorganismos tiene capacidad de causar dicha patología oncológica, por lo cual ha sido clasificados como un carcinógeno tipo 119,48.
La enfermedad presenta una prevalencia mundial cercana a los 900 000 individuos, según la OMS18. En el caso de Colombia, la liga contra el cáncer, estimaba más de 8700 casos nuevos así como unos 6630 decesos debidos a esta enfermedad. La prevalencia del cáncer en Colombia es del 9,3%,en donde el 96,9% eran adenocarcinomas y el 3,1% eran linfomas tipo MALT. La región con mayor prevalencia de cáncer gástrico fue la Región Andina de 9,7% en comparación con la Costa Atlántica con 6,7%9.
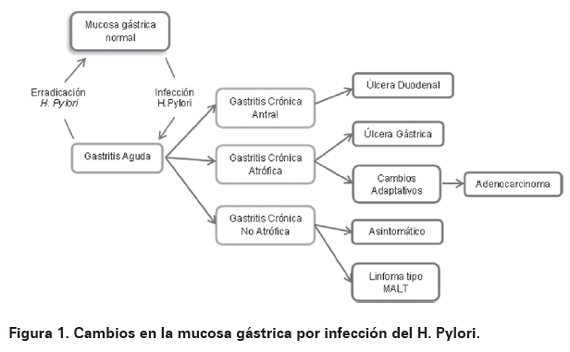
Sin embargo no es solo la infección por cepas del microorganismo vacA y cagA, sino que además la suceptibilidad del huésped es lo que determina el desarrollo de cáncer por lo que no todos los infectados desarrollan tal patología39. Ahora bien el desarrollo de las patologías asociadas a la infección por el microorganismo se da usualmente por cambios secuenciales de la mucosa19 que se resumen en la Figura 1. En el caso de la infección por H. pylori, el tipo de cáncer no solo se ubica en la región antral del estómago, sino que además es de variante intestinal18,71.
Linfoma tipo MALT: El Tejido Linfático Asociado a Mucosas o MALT, es un cúmulo no encapsulado pero delimitado de linfocitos, sobretodo linfocitos B. Adicionalmente estás agrupaciones se localizan en la mucosa del sistema digestivo, aunque también se hallan presentes en el sistema respiratorio y urinario72. Cuando está presente la infección por el H. pylori, se inicia la activación de la respuesta inmune secundaria a diferentes antígenos46,73-75, por lo que dentro de los factores de riesgo relacionados tanto con el adenocarcinoma como el linfoma tipo MALT, está la presencia de cepas positivas para el gen cagA. Los estudios han demostrado que este gen interactúa con los linfocitos B, uniéndose con receptores de tirosin-kinasa estimulando su activación46,59, a la vez que frena la apoptosis y favorece la conversión linfocítica así como el desarrollo de linfoma tipo MALT, el cual puede variar su grado de malignidad59. El tratamiento de la infección de la bacteria, ha demostrado ser efectivo para la regresión de linfoma tipo MALT en estadios iniciales.
Deficiencia de hierro si bien no hay estudios que confirmen totalmente esta hipótesis, se cree que el H. pylori puede ser u factor de riesgo para el desarrollo de anemia ferropénica48. Las manifestaciones clínicas pueden ir desde cansancio generalizado, piel pálida, hasta la pérdida crítica de peso. Una de las principales hipótesis que justifican esta relación es la pérdida de sangre, secundaria a úlceras duodenales que puedan o no sangrar; otra teoría es la captación del hierro por parte de la bacteria, ya que este es un micronutriente esencial; sin embargo esto parece estar más relacionado con cepas que tienen una sobreexpresión del producto del gen pfr, es decir, tienen una alta producción de ferritina con lo cual captaría y almacenaría más hierro para su supervivencia76,77. También han relacionado el producto del gen feoB que se ha identificado como un transportador de hierro en estado ferroso78, pese a que en las cepas estudiadas existe un alto polimorfismo de los genes implicados estudiados (pfr y feoB), la población ha sido muy poca y muy heterogénea, así como también que la anemia por deficiencia de hierro puede ser multifactorial76,78,79.
CONCLUSIONES
El estómago era considerado un estéril, pero esta idea fue replanteada en 1983 cuando los doctores Warren y Marshall reportaron la existencia de una bacteria causante de patologías estomacales. La bacteria inicialmente se denominó Campylobacter pylori, y tras secuenciar su genoma, lo renombraron como H. Pylori. Esta bacteria ha sido objeto de controversias y estudios, que han permitido dilucidar su papel, tanto fisiológico como patológico. Desde lo fisiológico, se resalta el aumento en la producción de ión amonio que actúa como tampón disminuyendo el pH estomacal previniendo el daño sobre la unión gastroesofágica, así como también favorece el aumento de la presión del esfínter esofágico inferior, evitando el reflujo y con ello el subsecuente daño sobre el epitelio del esófago. El aspecto patológico es mucho más amplio y va desde el desarrollo de gastritis agudas, hasta el desarrollo neoplasias malignas como el linfoma tipo MALT o el adenocarcinoma los cuales, sin un tratamiento adecuado pueden comprometer la vida de la persona. Otro de los grandes atractivos, al estudiar este microorganismo, es lo referente al papel desempeñado de los mecanismos inmunitarios celulares y humorales presentes durante la infección, y como estos son determinantes de la evolución de la enfermedad. También existe un gran interés por el estudio la genética bacteriana, tanto así se ha logrado secuenciar completamente el genoma del H. pylori, y gracias a esos estudios se han identificado dos tipos de bacterias que infectan el epitelio estomacal, así como también se identificaron genes e islas de patogenicidad responsables de convertir una cepa inocua, en una cepa agresiva para el epitelio. Sin duda alguna continuará las investigaciones en este campo, contribuyendo así al incremento en el conocimiento sobre la bacteria, su relación con el epitelio, sus potenciales beneficios así como sus potenciales procesos patológicos.
AGRADECIMIENTOS
Los autores agradecen al Dr. Julio Cesar Mantilla, médico patólogo de la UIS por la colaboración en la revisión previa del artículo.
REFERENCIAS BIBLIOGRÁFICAS
1. Sierra Arango, F. and D.d.P. Torres Pabón, Helicobacter pylori: El holocausto revolucionario. Ediciones médicas Latinoamericanas, 2001. [ Links ]
2. Flemming, S., Deadly Diseases and Epidemics: Helocibacter pylori. 2007, New York: Chelsea House Plublishers. [ Links ]
3. Ricaurte Guerrero, O., Patología de la infección por Helicobacter pylori. Rev Fac Med UN Col. 1997;45(1):32-39. [ Links ]
4. Majalca Martínez, C., et al., Transporte, aislamiento, identificación y conservación de cepas de Helicobacter pylori. Bioquimia. 2001;26(4):85-89. [ Links ]
5. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539-47. [ Links ]
6. Antonio-Rincón F, López Vidal Y, Castillo-Rojas G, Lazcano-Ponce EC, Ponce-de-León S, Tabche-Barrera ML, Aguilar-Gutiérrez GR. Pathogenicity island cag, vacA and IS605 genotypes in Mexican strains of Helicobacter pylori associated with peptic ulcers. Ann Clin Microbiol Antimicrob. 2011;10:18. [ Links ]
7. Hold GL, Mukhopadhya I, Monie TP. Innate immune sensors and gastrointestinal bacterial infections. Clin Dev Immunol. 2011:579-650. [ Links ]
8. Kennemann L, Didelot X, Aebischer T, Kuhn S, Drescher B, Droege B, et al. Helicobacter pylori genome evolution during human infection. Proc. Natl. Acad. Sci. U. S. A. 2011;108(12):5033-8. [ Links ]
9. Bravo LE, Cortés A, Carrascal E, Jaramillo R, García LS, Bravo PC, et al. Helicobacter pylori: patología y prevalencia en biopsias gástricas en Colombia. Colomb. med. 2003;34(3):124-31. [ Links ]
10. Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119(9):2475-87. [ Links ]
11. Gutierez O, Aponte D, Páramo D, Sabbagh LC, Ángel LA, Cardona H, et al. Seroprevalencia y factores de riesgo asociados con la infección de Helicobacter pylori. Rev colomb gastroenterol. 2000;16(1):19-22. [ Links ]
12. Warren JR, Marshall B. Unidentified cuved bacilli on gastric epithelium and active chronic gastritis. Lancet. 1983 Jun 4;321(8336):1273â5. [ Links ]
13. Alm RA, Bina J, Andrews BM, Doig P, Handcock RE, Trust TJ. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect Immun. 2000 Jul;68(7):4155-68. [ Links ]
14. Dong QJ, Wang Q, Xin YN, Li N, Xuan SY. Comparative genomics of Helicobacter pylori. World J Gastroenterol. 2009 Aug 28;15(32):3984-91. [ Links ]
15. Kauser F, Hussain MA, Ahmed I, Srinivas S, Devi SM, Majeed AA, et al. Comparative genomics of Helicobacter pylori isolates recovered from ulcer disease patients in England. BMC Microbiol. 2005 May 25;5:32. [ Links ]
16. Ahmed N, Majeed AA, Ahmed I, Hussain MA, Alvi A, Devi SM, et al. genoBASE pylori: a genotype search tool and database of the human gastric pathogen Helicobacter pylori. Infect Genet Evol. 2007 Jul;7(4):463-8. [ Links ]
17. de-Reuse H, Bereswill S. Ten years after the first Helicobacter pylori genome: comparative and functional genomics provide new insights in the variability and adaptability of a persistent pathogen. FEMS Immunol Med Microbiol. 2007 Jul;50(2):165-76. [ Links ]
18. Fuentes-Pananá E, Carmolinga-Ponce M, Maldonado-Bernal C. Infección, inflamación y cáncer gástrico. Salud pública Méx. 2009;51(5):427-33. [ Links ]
19. Romo-González C, Coria-Jiménez CR. Helicobacter pylori, un modelo de bacteria carcinogénica. Rev Esp Med Quir. 2010;15(4):242-51. [ Links ]
20. Mendall MA, Northfield TC. Transmission of Helicobacter pylori infection. Gut. 1995;37(1):1-3. [ Links ]
21. Bedoya A, Garay J, Sansón F, Bravo LE, Correa H, et al. Histopathology of gastritis in Helicobacter pylori-infected children from populations at high and low gastric cancer risk. Hum Pathol. 2003;34(3):206-13. [ Links ]
22. Bedoya A, Arcos M, Sansón F, Del-Castillo G. Helicobacter pylori y cambios histológicos de la mucosa gástrica en menores de diez años: pasto, 1999. Rev colomb gastroenterol. 2002 Mar;17(1):36-41. [ Links ]
23. Gutiérrez O, Gómez M, Castillo de Moreno M. La infección gástrica por helicobacter pylori modifica la secreción de ácido. Rev Fac Med UN Col. 2001;49(2):76-80. [ Links ]
24. Cittelly DM, Huertas MG, Martínez JD, Oliveros R, Posso H, Bravo MM, Orozco O. Helicobacter pylori genotypes in nonatrophic gastritis are different of the found in peptic ulcer, premalignant lesions and gastric cancer in Colombia. Rev Med Chil. 2002 Feb;130(2):143-51. [ Links ]
25. Cittelly DM, Gutierrez O, Huertas M, Martínez J, Oliveros R, Posso H, et al. Genotipos cagA, vacA e iceA de aislamientos colombianos de Helicobacter pylori. Rev colomb gastroenterol. 2002 Jun;17(2):99-105. [ Links ]
26. Telford JL, Ghiara P, DellàOrco M, Comanducci M, Burroni D, Bugnoli M, et al. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J Exp Med. 1994 May;179(5):1653-58. [ Links ]
27. Ortiz-Princz D, Guariglia-Oropeza V, Ávila M, Correnti M, Perrone M, Gutierrez B, Torres J, et al. Helicobacter pylori cagA and vacA genotypes in Cuban and Venezuelan populations. Mem Inst Oswaldo Cruz. 2010 May;105(3):331-5. [ Links ]
28. Saxena A, Shukla S, Prasad KN, Ghoshal UC. Virulence attributes of Helicobacter pylori isolates & their association with gastroduodenal disease. Indian J Med Res. 2011;133(5):514-20. [ Links ]
29. Yamaoka Y. Increasing evidence of the role of Helicobacter pylori SabA in the pathogenesis of gastroduodenal disease. J Infect Dev Ctries. 2008;2(3):174-81. [ Links ]
30. Mahdavi J, Sondén B, Hurting M, Olfat FO, Roche N, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002 Jul;297(5581):573-8. [ Links ]
31. Pride DT, Meinersmann RJ, Blaser MJ. Allelic Variation within Helicobacter pylori babA and babB. Infect Immun. 2001;69(2):1160-71. [ Links ]
32. Yamaoka Y. Roles of Helicobacter pylori BabA in gastroduodenal pathogenesis. World J Gastroenterol. 2008;14(27):4265-72. [ Links ]
33. Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol. 2010 Nov. 7(11): 629-41. [ Links ]
34. Issa S, Moran AP, Ustinow SN, Lin JH, Ligtenberg AJ, Karlsson NG. O-linked oligosaccharides from salivary agglutinin: Helicobacter pylori binding sialyl-Lewis x and Lewis b are terminating moieties on hyperfucosylated oligo-N-acetyllactosamine. Glycobiology. 2010 Aug;20(8):1046-57. [ Links ]
35. Linden S, et al. Role of mucin Lewis status in resistance to Helicobacter pylori infection in pediatric patients. Helicobacter. 2010;15(4):251-8. [ Links ]
36. Cappon A, Babolin C, Segat D, Cancian L, Amedei A, Calzett F, et al. Helicobacter pylori-derived neutrophil-activating protein increases the lifespan of monocytes and neutrophils. Cell Microbiol. 2010 Jun;12(6):754-64. [ Links ]
37. Garza-Gonzalez E, Pérez-Pérez FI, Tijerina-Menchaca R, Maldonado-Garza HJ, Bosques-Padilla FJ. Helicobacter pylori genotypes and their association with hostàs immune response. Rev Gastroenterol Mex. 2002;67(3):155-60. [ Links ]
38. Park, S.M, et al. Relevance of vacA genotypes of Helicobacter pylori to cagA status and its clinical outcome. Korean J Intern Med, 2001;16(1):8-13. [ Links ]
39. Yin, M, et al. Molecular epidemiology of genetic susceptibility to gastric cancer: focus on single nucleotide polymorphisms in gastric carcinogenesis. Am J Transl Res. 2009;1(1):44-54. [ Links ]
40. Do Carmo, AP, Rabenhorst SH. Importance of vacAs1 gene in gastric cancer patients infected with cagA-negative Helicobacter pylori. APMIS. 2011;119(7):485-6. [ Links ]
41. Toller IM, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci U S A. 2011;108(36):14944-9. [ Links ]
42. Ahmad, T, et al. Prevalence of Helicobacter pylori pathogenicity-associated cagA and vacA genotypes among Pakistani dyspeptic patients. FEMS Immunol Med Microbiol. 2009;55(1):34-8. [ Links ]
43. Toller IM, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci U S A. 108(36):14944-9. [ Links ]
44. Boonyanugomol W, et al. Role of cagA-Positive Helicobacter pylori on Cell Proliferation, Apoptosis, and Inflammation in Biliary Cells. Dig Dis Sci. 2010;56(6):1682-92. [ Links ]
45. Rosebeck S, Lucas PC, McAllister-Lucas LM. Protease activity of the API2-MALT1 fusion oncoprotein in MALT lymphoma development and treatment. Future Oncol. 2011;7(5):613-7. [ Links ]
46. Lin WC, et al. Translocation of Helicobacter pylori CagA into Human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer Res. 2010;70(14):5740-8. [ Links ]
47. Munari, F, et al. Tumor-associated macrophages as major source of APRIL in gastric MALT lymphoma. Blood. 2011;117(24):6612-6. [ Links ]
48. McColl KE. Clinical practice. Helicobacter pylori infection. N Engl J Med. 2010;362(17):1597-604. [ Links ]
49. Cardona HJ, et al. ¿Es el Helicobacter pylori un factor protector para el desarrollo de reflujo gastroesofágico? Rev. colomb. Gastroenterol. 2002;17(1):13-9. [ Links ]
50. Dorer MS, Talarico S, Salama NR. Helicobacter pyloriàs unconventional role in health and disease. PLoS Pathog. 2009;5(10):p.e1000544. [ Links ]
51. Sharma P. Clinical practice. Barrettàs esophagus. N Engl J Med. 2009;361(26):2548-56. [ Links ]
52. Yaghoobi M, et al. Is there an increased risk of GERD after Helicobacter pylori eradication?: a meta-analysis. Am J Gastroenterol. 105(5):p.1007-13;quiz1006,1014. [ Links ]
53. Laine L, Sugg J. Effect of Helicobacter pylori eradication on development of erosive esophagitis and gastroesophageal reflux disease symptoms: a post hoc analysis of eight double blind prospective studies. Am J Gastroenterol. 2002; 97(12): 2992-7. [ Links ]
54. Kearney DJ, et al. The effect of a Helicobacter pylori treatment strategy on health care expenditures in patients with peptic ulcer disease and dyspepsia. Am J Gastroenterol. 2003;98(9):1952-62. [ Links ]
55. Abnet CC, et al. Plasma pepsinogens, antibodies against Helicobacter pylori, and risk of gastric cancer in the Shanghai Womenàs Health Study Cohort. Br J Cancer. 2011;104(9):1511-6. [ Links ]
56. Alves MK, et al. CDKN2A promoter methylation is related to the tumor location and histological subtype and associated with Helicobacter pylori flaA(+) strains in gastric adenocarcinomas. APMIS. 2010;118(4):297-307. [ Links ]
57. Andre AR, et al. Gastric adenocarcinoma and Helicobacter pylori: correlation with p53 mutation and p27 immunoexpression. Cancer Epidemiol. 2010;34(5):618-25. [ Links ]
58. Cho SO, et al. Involvement of Ras and AP-1 in Helicobacter pylori-induced expression of COX-2 and iNOS in gastric epithelial AGS cells. Dig Dis Sci. 2009;55(4):988-96. [ Links ]
59. Zalewska-Ziob M, et al. TNF-alpha expression in gastric mucosa of individuals infected with different virulent Helicobacter pylori strains. Med Sci Monit. 2009;15(6):p.BR166-71. [ Links ]
60. Osawa H. Ghrelin and Helicobacter pylori infection. World J Gastroenterol. 2008. 14(41): p. 6327-33. [ Links ]
61. Gutierrez CO. La infección gástrica por helicobacter pylori modifica la secreción gástrica de ácido. Rev Fac Med UN Col. 2001;49(2):76-80. [ Links ]
62. Beswick EJ, Reyes VE. CD74 in antigen presentation, inflammation, and cancers of the gastrointestinal tract. World J Gastroenterol. 2009;15(23):2855-61. [ Links ]
63. Takeshima E, et al. Helicobacter pylori-induced interleukin-12 p40 expression. Infect Immun. 2009;77(4):1337-48. [ Links ]
64. Azuma T, et al. The role of the HLA-DQA1 gene in resistance to atrophic gastritis and gastric adenocarcinoma induced by Helicobacter pylori infection. Cancer. 1998;82(6):1013-8. [ Links ]
65. Perri F, et al. HLA-DQA1 and -DQB1 genes and Helicobacter pylori infection in Italian patients with gastric adenocarcinoma. Tissue Antigens. 2002;59(1):55-7. [ Links ]
66. Watanabe Y, et al. HLA-DQB1 locus and gastric cancer in Helicobacter pylori infection. J Gastroenterol Hepatol. 2006;21(2):420-4. [ Links ]
67. Kumar V, Abbas AK, Fausto N. Estómago, in Robbins y Cotran: Patología estructural y funcional. Elsevier: Madrid. 2005;p.815-31. [ Links ]
68. Rubin E, et al. El aparato digestivo., in Patología estructural: fundamentos clinicopatológicos en medicina. McGraw Hill: Barcelona. 2006;619-30. [ Links ]
69. Andres S, et al. Helicobacter pylori defines local immune response through interaction with dendritic cells. FEMS Immunol Med Microbiol. 2010;61(2):168-78. [ Links ]
70. Zhernakova NI. Clinical manifestations of Helicobacter pylori-induced ulcer disease in elderly patients. Klin Med (Mosk). 2009;87(4):44-6. [ Links ]
71. Bai H, Li Q, Liu X, Li Y. Characteristics and interactions of Helicobacter pylori and H. pylori-infected human gastroduodenal epithelium in peptic ulcer: a transmission electron microscopy study. Dig Dis Sci. 2009;55(1):82-8. [ Links ]
72. Gartner LP, Hiatt JL. Texto atlas de histología. Segunda ed. McGraw Hill. México. 2001;p.285. [ Links ]
73. Tsai HF, Hsu PN. Interplay between Helicobacter pylori and immune cells in immune pathogenesis of gastric inflammation and mucosal pathology. Cell Mol Immunol. 2010;7(4):255-9. [ Links ]
74. Zullo A, Hassan C, Cristofari F, Andriani A, De Francesco V, Ierardi E, et al. Effects of Helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin Gastroenterol Hepatol. 2009;8(2):105-10. [ Links ]
75. Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet. 1991;338(8776):1175-6. [ Links ]
76. Choe YH, Hwang TS, Kim HJ, Shin SH, Song SU, Choi MS. A possible relation of the Helicobacter pylori pfr gene to iron deficiency anemia? Helicobacter. 2001;6(1):55-9. [ Links ]
77. Waidner B, Greiner S, Odenbreit S, Kavermann H, Velayudhan J, Stähler F, et al. Essential role of ferritin Pfr in Helicobacter pylori iron metabolism and gastric colonization. Infect Immun. 2002;70(7):3923-9. [ Links ]
78. Jeon BH, Oh YJ, Lee NG, Choe YH. Polymorphism of the Helicobacter pylori feoB gene in Korea: a possible relation with iron-deficiency anemia? Helicobacter. 2004;9(4):330-4. [ Links ]
79. Fernández-Bañares F, Monzón H, Forné M. A short review of malabsorption and anemia. World J Gastroenterol. 2009;15(37):4644-52. [ Links ]
