










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.25 no.1 Bicaramanga Jan./Apr. 2012
Síndrome de Kabuki: caracterización clínica,
estudios genéticos, manejo preventivo de las
complicaciones y asesoría genética
Jorge Luis Suárez-Guerrero*
Gustavo Adolfo Contreras-García**
*Estudiante VI semestre. Escuela de Medicina. Miembro SEIMED. Grupo de investigación en genética clínica. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
**MD Genetista. Docente - Coordinador del Área de Investigación en Genética Clínica. Grupo de Genética Humana. Universidad Industrial de Santander. Departamento de Pediatría. Hospital Universitario de Santander. Bucaramanga. Santander. Colombia.
Correspondencia: Sr. Suarez. Calle 4 #12-56 Nuevo Villabel. Floridablanca. Santander. Colombia. e-mail: jorgesuarezg@gmail.com; jorgesuarezg_@hotmail.com.
Artículo recibido el 29 de agosto de 2011 y aceptado para publicación el 20 de abril de 2012.
RESUMEN
Introducción: Kabuki hace referencia al teatro tradicional japonés, y el nombre del síndrome proviene de la semejanza de los pacientes al maquillaje facial usado por dichos actores. El síndrome de Kabuki es una patología dismorfológica, caracterizada por rasgos faciales particulares, entre ellos fisuras palpebrales amplias, puente nasal deprimido, cejas arqueadas con el tercio externo disperso y orejas prominentes. Todos estos rasgos son concomitantes con retraso mental, cardiopatías, nefropatías, entre otros. Debido a la presencia en mayor o menor número de veces de algunas patologías, se han dividido en anomalías menores y anomalías mayores. Objetivos: Presentar una revisión sobre las generalidades del síndrome de Kabuki, características clínicas, complicaciones y su manejo preventivo, así como los estudios genéticos realizados hasta la fecha y la asesoría genética. Metodología se utilizaron las bases de datos Pubmed y SciELO, para la búsqueda de información Resultados: se encontraron estudios publicados alusivos a los primeros casos del síndrome, hasta aquellos recientemente publicados en donde se identifica el gen MLL2 como etiología para este síndrome. Conclusiones: hasta la fecha, el diagnóstico se realiza por los hallazgos clínicos, aunque se puede detectar la mutación del gen MLL2. Para el diagnóstico se tienen presente los antecedentes familiares y los hallazgos al examen físico, principalmente los rasgos faciales propios de este síndrome. Complementando el diagnóstico, se debe llevar a cabo un manejo preventivo de las complicaciones para así evitar potenciales riesgos, además de ofrecer a la familia información necesaria durante la asesoría genética. (MÉD.UIS. 2011;25(1):19-27).
Palabras Clave: Síndrome de Kabuki. Anomalías craneofaciales. Cejas arqueadas. Cejas dispersas. Fisuras palpebrales largas. Labio fisurado. Fisura palatina.
ABSTRACT
Kabuki syndrome: clinical, genetic, preventive management of complications and genetic counseling
Introduction: Kabuki refers to traditional Japanese theater, and the syndrome's name comes from the similarity of the patients' facial makeup used by these actors. Kabuki syndrome is a dismorfological pathology characterized by particular facial features including wide palpebral fissures, depressed nasal tip, arched eyebrows with the lateral one-third dispersed or sparse, and prominent ears. All these features are concomitant, with mental retardation, cardiopathies, nephropathies , among others. Due to the presence in greater or lesser number of times certain pathologies, have been divided into minor and major abnormalities. Objective: present a review of the generalities of Kabuki syndrome, dismorfologicas features, clinical characteristics, complications, and genetic studies to date. Methods: we used the databases PubMed and SciELO, to search for information. Results: the published studies alluding to the first cases of the syndrome, even those published recently where MLL2 gene is identified as a possible candidate for this syndrome. Conclusions: until now the diagnosis is made by clinical findings, although it can detect the mutation of gene MLL2. For the diagnosis is given through the family history and physical examination findings, especially the facial features, characteristic of this syndrome. Complementing the diagnosis must be carry out a preventive management of complications and to avoid potential risks, and offer the family information during genetic counseling. (MÉD.UIS. 2011;25(1):19-27)
Keywords: Kabuki Syndrome. Craniofacial anomalies. Arched eyebrows. Sparse eyebrows. Long palpebral fissures. Cleft lip/palate.
INTRODUCCIÓN
Originalmente descrito de forma independiente en 1981 por los doctores Niikawa1 y Kuroki2-6, el síndrome de Kabuki, KMS (OMIM #147920), síndrome maquillaje de Kabuki o síndrome Niikawa-Kuroki, presenta múltiples anomalías faciales asociadas en la mayoría de los casos con retardo mental, que puede variar entre leve a severo. Kabuki es el nombre del teatro tradicional japonés en donde sus actores utilizan un maquillaje particular, que consiste en base color blanco y delineación arqueada en color negro de las cejas, entre otros aspectos1,7. Debido a su semejanza, Niikawa1 asignó este nombre a las personas que padecían este síndrome. En la actualidad, el término "maquillaje" tiende a su desuso porque se considera un término despectivo4, 5, 8.
Los primeros 62 reportes se realizaron en 1988 mediante el trabajo de Niikawa et al9, quienes además de informar los casos, instauraron las bases del diagnóstico de KMS según cinco condiciones principales:
- Rasgos faciales.
- Anomalías esqueléticas.
- Alteración en los dermatoglifos.
- Retardo mental de tipo leve a moderado.
- Baja talla para la edad.
De todos ellos, han sido los rasgos faciales los más usados para su identificación. Con base en los hallazgos de Niikawa y Kuroki, Matsumoto et al8 propusieron una nueva organización de estos cinco rasgos en dos grandes categorías: rasgos mayores y rasgos menores. Dentro de los rasgos mayores, se incluyen: fisuras palpebrales amplias, eversión del parpado inferior, puente nasal deprimido, cejas arqueadas, pabellones auriculares prominentes o malformados, paladar alto o hendido, dentadura anormal, persistencia de almohadillas en el pulpejo de los dedos, clinodactilia del quinto dedo o braquidactilia, hipotonía, retardo mental, baja estatura, hipoacusia y ptosis palpebral1,2,4,6,9-14. Los rasgos menores son: escleras azules, escoliosis, cardiopatías, nefropatías, vértebras malformadas, criptorquidia, deficiencia de la hormona del crecimiento, entre otros1, 2, 4, 6, 9-14.
Tras la publicación de los primeros casos en Japón, el número de reportes a nivel mundial ha aumentado, tanto así que hoy día se han logrado divulgar más de 400 casos en todo el mundo4,8,11,12,14-31. Para América Latina, las publicaciones han aumentado considerablemente en los últimos años32-41. Esta información, ha permitido demostrar que hombres y mujeres se afectan de manera similar en diferentes razas y regiones geográficas4,10. Se ha estimado una prevalencia del KMS de 1 en 32 0002,9,10,29,42 en población japonesa y se han reportado casos de individuos afectados en otros países no asiáticos, por ejemplo en Oceanía con prevalencia de 1 en 86 000 nacidos vivos2,42,43. Se han publicado algunas diferencias menores entre pacientes asiáticos y no asiáticos, sin embargo los rasgos principales son comunes en todas las poblaciones4,5. A pesar de la bibliografía disponible, este síndrome aún es desconocido por muchos profesionales del área de la salud debido a su reciente descripción, generando con ello un posible subregistro de casos en la población.
METODOLOGIA DE BUSQUEDA
Como método de búsqueda de la información relacionada con el síndrome de Kabuki, se llevó a cabo una investigación sistemática en las bases de datos PubMed y SciELO. Para la búsqueda en Pubmed se emplearon los términos: "Niikawa-Kuroki syndrome", "Kabuki make up syndrome" y "Kabuki syndrome", todos pertenecientes a MeSH. Por su parte para la búsqueda en SciELO, se utilizaron los términos "Kabuki" y "Síndrome de Kabuki". Para la organización y acceso a la información bibliográfica se empleó el manejador de referencias endnote-X4®.
CARACTERÍSTICAS CLÍNNICAS
Teniendo presente que su diagnóstico es clínico, el personal de la salud en general y los médicos en particular deberían tener presente los rasgos fenotípicos más frecuentes (Ver Tabla 1) y sobresalientes como son:
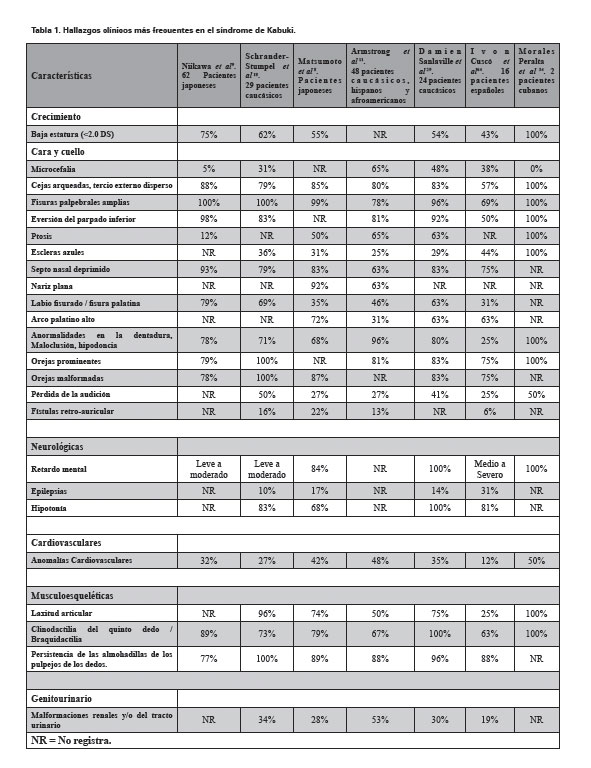
A nivel cráneo-facial: las fisuras palpebrales amplias, eversión del párpado inferior, cejas arqueadas con el tercio externo disperso (Ver Figura 1), puente nasal deprimido, punta de la nariz plana, paladar fisurado o arco palatino alto, pabellones auriculares prominentes o malformados, ptosis palpebral y escleras azules4,5, hipodoncia o dentadura anormal5,7. El promedio del perímetro cefálico en la mayoría de los pacientes reportados está en el rango normal. Sin embargo, como algunos casos presentan microcefalia, se recomienda vigilar este parámetro4.
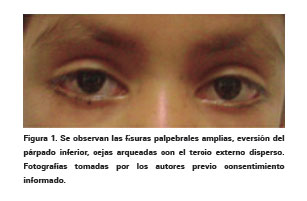
En manos: persistencia de la almohadilla en el pulpejo de los dedos hasta en un 95% de los casos (Ver Figura 2), clinodactilia del quinto dedo y braquidactilia de los dedos de la mano2,4,11,14,42.
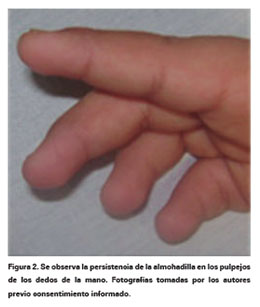
Desde el punto de vista neurológico: la mayoría de los pacientes presentan compromiso neurológico teniendo diferente expresión clínica ya que las manifestaciones son variables, entre estas se encuentran hipotonía, retraso psicomotor que puede terminar en retardo mental y este teniendo en cuenta el coeficiente intelectual puede ser leve, moderado o severo4,9,12,42,44,45. En algunos casos se ha reportado hipotonía12,42,44. Existen también descripciones de casos sin retardo mental pero con trastornos conductuales, así como paciente con el resto de criterios clínicos pero neurológicamente normales que representaría un bajo porcentaje8,46 (Ver Tabla 1).
Con respecto al crecimiento: las personas con el síndrome pueden tener una baja talla2,4,8,9,14,20, usualmente por debajo del percentil 5 para la edad.
En sistema auditivo: se ha demostrado hipoacusia conductiva generalmente secundaria a otitis media a repetición, en la mayor parte de los casos progresiva42, 44, 47. De igual forma se han reportado pacientes con hipoacusia neurosensorial47,48 así como hipoacusia mixta, es decir neurosensorial y conductiva47.
A nivel cardiovascular: se han identificado malformaciones como coartación de aorta15,19, doble arco aórtico49, defectos del septo ventricular15 y aneurismas50, entre otros.
Sistema renal: se han reportado malformaciones como agenesia renal unilateral51 y megauréter52, entre otros.
HALLAZGOS GENÉTICOS
Si bien han sido múltiples los estudios moleculares que se han hecho, solo hasta hace poco se identificó la causa del síndrome de Kabuki53-56. Con respecto al cariotipo mediante bandeo G de alta resolución que es la prueba de mayor uso en clínica, esta se ha encontrado normal en la mayoría de los casos5,29; sin embargo, por la metodología empleada no se puede descartar la presencia de pequeños rearreglos cromosómicos o bajos niveles de mosaicismo, es precisamente esto lo que ha llevado al uso de técnicas mucho más complejas. La mayoría de casos se han reportado de manera esporádica en las familias, lo que plantea como principal alteración una mutación de novo, siendo compatible principalmente con un mecanismo de herencia autosomico dominante10,57-9.
En lo referente a las pruebas moleculares, los primeros estudios adelantados por investigadores como Milunsky et al60, 61 en el año 2003, quienes mediante el uso de la Hibridación Genómica Comparativa o CGH por sus siglas en inglés, y la Hibridación in Situ con Fluorescencia o FISH, reportaron una duplicación de 3,5 Mb submicroscópica del cromosoma 8 en 8p22-8p23.1 en seis de las personas del estudio. Las siguientes investigaciones fueron llevadas a cabo en el año 2005 por Shieh et al62 quienes reportaron la presencia de una triplicación de las regiones 8p22-8p23, en una paciente con fenotipo similar a Kabuki.
Sin embargo, trabajos independientes llevados a cabo por MIyake et al63 en el año 2004, Schouman et al28 y Sanlaville et al29 en el 2005, Kimberly et al31 y Cuscó et al64 en el 2006, manifestaron que en sus estudios no detectaron duplicación, inversión o deleción alguna en 8p22-8p23.1, contrastando de este modo con los hallazgos de Milunsky y los de Shieh. Por su parte, Cuscó et al64 además de no haber encontrado hallazgos en el cromosoma 8, sí reportó que cinco de las personas en el estudio presentaron variaciones del material genético en los cromosomas 2, 5, 14, 16 y 17. Para el año 2007, Maas et al10, identificaron en una niña una mutación de novo, en el exón 5 del gen C20orf133 en el cromosoma 20p12.1, pero esta mutación no fue detectada en otros pacientes. Kuniba et al65, describen en su trabajo la genotipificación en 17 pacientes con KMS se obtuvo una región candidata en 9q21 en la cual se encuentra cuatro genes que pudieran relacionarse con el síndrome: TRPM3, KLF9, SMC5 y MAMDC2. De estos genes, el TRPM3 (Transient Receptor Potential Cation Channel, subfamily M, member 3) en 9q21.11-q21.12, se relacionó anteriormente con labio/paladar hendido, por lo que podría relacionarse a esta característica.
En fechas recientes, investigaciones independientes empleando la técnica de secuencia de exomas, identificaron alteraciones en el gen MLL2 y lo relacionaron como causa de este síndrome, la alteración de dicho gen se ha encontrado en la mayoría de los pacientes con este síndrome, sin embargo por no ser en el 100% se considera que puede existir heterogeneidad de loci, lo que explicaría este resultado. Una de las primeras investigaciones fue llevada a cabo por Sarah Ng et al53, quienes detectaron la mutación en nueve de diez personas. Por su parte, Paulussen et al56 la identificaron en 34 de 45 pacientes, Micale et al54 la hallaron en 45 de 62 pacientes, así como Banka et al55 la identificaron en 74 de 116 pacientes. Sin duda, la mutación de este gen, que pertenece a la familia de las metiltransferasas, es la principal causa para el desarrollo de KMS, ya que su alteración afecta los procesos de la transcripción de diversos genes durante embriogénesis, explicando con ello las características faciales y las malformaciones en diferentes órganos55.
COMPLICACIONES
Las complicaciones derivadas de este síndrome son otro de los temas a tener presente durante la consulta. Sin duda alguna, el retardo mental es una condición que empeora el cuadro entre mayor sea, lo que puede conllevar a una menor capacidad de la persona para tener un estilo de vida independiente. En cuanto a las malformaciones de la cavidad orofaríngea, estas pueden ser desde anomalías menores manejables sin mayores complicaciones, hasta dificultar la alimentación con necesidad de sonda nasogástrica o incluso gastrostomía. La hipoacusia se puede presentar hasta en el 40-45% de las personas con el síndrome42,44,47, bien sea una hipoacusia conductiva secundaria a otitis media crónica44,47, por una hipoacusa neurosensorial48 o por afección tanto conductiva como neurosensorial47. En muchos casos las malformaciones facilitan la aparición de neumonías por aspiración5,7,10,11. La fisura palatina también puede alterar el desarrollo del lenguaje, que sumado a la hipoacusia o sordera, dependiendo de cómo evolucione el daño neuronal, desmejora notablemente las habilidades comunicativas de la persona. Las malformaciones renales también suelen presentarse10, así como las cardiopatías cuyos valores de presentación oscilan entre el 32 y el 58% de los casos33,58,66 y de no ser tratadas a tiempo, pueden ser fatales.
Cabe informar que muchos de los pacientes con Kabuki pueden presentar con menor frecuencia otras anormalidades, entre ellas malformaciones como el ductus arterioso, coartación de aórtica5,12,42,49, hernias diafragmáticas, nefropatías, alteraciones de la vesícula biliar4, escoliosis, laxitud aumentada en los tendones, dislocación de la cadera33 e incluso alteraciones en el cordón umbilical en donde solo haya una arteria y una vena lo cual podría tener complicaciones in utero5. Se ha reportado la presencia de glaucoma, así como de otras anormalidades de la visión, sin embargo hacen falta más datos para confirmar esta relación11,34. Debido a la importancia de estas patologías, deben llevarse a cabo evaluaciones semiológicas y exámenes más cuidadosos para su diagnóstico temprano, cambiando de esta forma, no solo la estadística y los reportes de casos, sino el tratamiento oportuno y con ello, la calidad y expectativa de vida.
MANEJO PREVENTIVO DE LAS COMPLICACIONES
El tamizaje y el manejo médico deben ser oportunos e interdisciplinarios, indicando con esto que se deben tener seguimiento a los pacientes desde neonatología, cuando existe sospecha de alguna alteración e incluso más sabiendo que este síndrome puede estar acompañado de otras patologías36,67-70 haciendo su manejo bastante complejo. Además de ello, es importante hacer énfasis del soporte familiar en todo el proceso por ser de vital importancia. La guía de manejo clínico desarrollada por Dyscerne y Nowgene44 y en "Management of genetic syndromes"42 recomiendan que, tanto en la infancia como en la adolescencia, se lleven a cabo las pruebas diagnósticas que permitan descartar o confirmar la malformación de órganos como el corazón y los riñones, entre otros, para adelantar así las respectivas correcciones sean quirúrgicas o no. Para el sistema genitourinario, se deben descartar anomalías renales, de los uréteres, la vejiga y demás órganos relacionados por lo cual es recomendado los exámenes de ultrasonido renal y orina, entre otros.
En cuanto al sistema cardiovascular, por ser uno de lo más comprometidos15,58,71,72, una vez confirmado el síndrome, se deben llevar a cabo obligatoriamente un ecocardiograma en todas las personas en busca de patologías como coartación de la aorta42,49,50 y defectos del septo ventricular o auricular, entre otras; si se confirma alguna de estas patologías se recomienda seguimiento anual. El electrocardiograma no se considera un estudio de rutina en estos pacientes, aunque se debe realizar si se tienen hallazgos clínicos de arritmia. El tratamiento y monitoreo adelantado por cardiología, debe complementarse en muchas ocasiones con intervenciones quirúrgicas bien sea para corrección del defecto o para el trasplante del órgano. Acompañando al sistema cardiovascular está el respiratorio, quien se ve frecuentemente afectado por infecciones y es por ello que el monitoreo a cargo del especialista es de suma importancia, principalmente si se le han diagnosticado infecciones respiratorias previas. En el caso del sistema gastrointestinal es importante identificar si existen problemas de alimentación, de reflujo o alguna malformación del sistema.
Desde el aspecto neurológico es importante identificar desde edades tempranas el grado de afección que puede manifestarse bien sea por crisis epilépticas, afección neuromuscular o retardo mental, entre otras. En cuanto a las crisis epilépticas, debe hacerse el tratamiento y seguimiento por el especialista en neurología; cuando hay hipotonía las terapias físicas y de fortalecimiento muscular son las más indicadas. En el retardo mental, las habilidades tanto del comportamiento como intelectuales45, deben orientarse al desarrollo de destrezas sociales básicas como habilidades de la comunicación y la interacción con otras personas, siendo muy importante el trabajo con los familiares. En el aspecto académico, deben dirigirse los progresos acompañados de evaluaciones psicológicas escolares14 y observación de sus calificaciones, para mencionar algunos.
Es importante determinar la presencia de labio fisurado o fisura palatina, porque si persisten durante la infancia, la persona tendrá mayor dificultad al usar el lenguaje hablado. Por ello debe llevarse a cabo cirugías correctivas, trabajos con terapistas del lenguaje, así como con fonoaudiología y neurología, para detectar o descartar cualquier otra alteración que afecte de igual manera la adquisición del lenguaje. La pérdida de la audición, es una de las afecciones crónicas que aquejan a estos pacientes, es por ello que desde la infancia deben hacerse controles por parte del especialista, con el objetivo de evaluar la capacidad auditiva, descartar la displasia de Mondini, así como prevenir o tratar adecuadamente la otitis media42,44. Muchas veces el tratamiento no solo incluye el implante coclear sino un trabajo interdisciplinar liderado por el fonoaudiólogo y terapistas del lenguaje para mejorar sus habilidades comunicativas, incluso por especialistas en enfermedades infecciosas para el tratamiento de las otitis recurrentes. Es relevante tener presente las malformaciones y complicaciones de órganos menos comprometidos, como los ojos, en los cuales puede haber estrabismo, cataratas o glaucoma, por ello los chequeos por parte de oftalmología y optometría deben ser de forma periódica.
El sistema osteomuscular también se ve afectado, es por ello que se debe tener presente la hipotonía y la laxitud aumentada de ligamentos y tendones. Este aumento conlleva a una de las patologías más comunes en esta población que es la dislocación de cadera. Para todo ello se deben hacer trabajos por parte de fisioterapia y ortopedia. Los primeros encargados del mejoramiento de la fuerza muscular, la coordinación, el equilibrio y demás que sean necesarias para evitar las lesiones; los segundos deben encargarse del monitoreo en caso de sospecha, o realizar las correcciones necesarias cuando sean confirmadas las patologías, bien sea dislocación de cadera, escoliosis y dislocación de la patela, entre otras. El sistema endocrino, debe evaluarse principalmente por telarquia prematura, presente hasta en un 50% de las niñas, así como evaluar al paciente si presenta retardo en el crecimiento debido a deficiencias en la hormona del crecimiento o por hipotiroidismo42.
Si los padres no tienen criterios clínicos que sugieran el diagnóstico del síndrome, se debe considerar una mutación de novo, por lo que el riesgo de recurrencia sería menor al 1%. Por otro lado, el individuo afectado podría tener una probabilidad del 50% de transmitir la condición a la descendencia, debido a que se ha demostrado un mecanismo de herencia autosómico dominante. Finalmente cabe recalcar que todas las patologías presentes deben ser tratadas en la medida de lo posible por especialistas, pero mediante un trabajo interdisciplinar que debe estar dirigido por el Pediatra, Médico Internista o Genetista Clínico, con el fin de conseguir los mejores resultados y de esta forma mejorar en mayor medida la expectativa y calidad de vida de estas personas.
CONCLUSIONES
El KMS es una condición que se manifiesta por alteraciones faciales acompañadas de retardo mental así como complicaciones en otros órganos. Los primeros registros provienen de la década de los 80 cuando dos investigadores trabajando de forma independiente dieron las primeras luces sobre la existencia y caracterización de este síndrome. Hoy día si bien se conoce más acerca de esta patología y los reportes de caso han ido en aumento, tanto a nivel mundial como de Latinoamérica, es de esperar que aún sea desconocido por muchos de los trabajadores del área de la salud en general y médicos en particular. El diagnóstico temprano junto al trabajo interdisciplinar son de suma importancia para llevar a cabo un manejo preventivo y terapéutico apropiado, logrando con ello tener un panorama de las diferentes complicaciones de este síndrome para poder llevar a cabo una intervención prematura, favoreciendo con ello la salud integral de la persona y de esta forma mejorar en la medida de lo posible la calidad de vida de estas personas.
REFERENCIAS BIBLIOGRÁFICAS
1. Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a new syndrome of mental retardation, unusual facies, large and protruding ears, and posnatal growth deficiency. J Pediatr. 1981;99(4):565-9. [ Links ]
2. Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet. 2005;67(3): 209-19. [ Links ]
3. Lung ZH, Rennie A. Kabuki syndrome: a case report. J Orthod. 2006;33(4): 242-5. [ Links ]
4. Pascual-Castroviejo I, Pascual-Pascual SI, Velázquez-Fragua R, Palencia R. Kabuki make-up syndrome. A report of 18 Spanish cases. Rev Neurol. 2005;40(8):473-8. [ Links ]
5. Burke LW, Jones MC. Kabuki syndrome: underdiagnosed recognizable pattern in cleft palate patients. Cleft Palate Craniofac J. 1995;32(1):77-84. [ Links ]
6. Kuroki Y, Suzuki Y, Chyi H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99(4):570-3. [ Links ]
7. Kobayashi ET, Maruyama Y, Kobayashi K. A longitudinal evaluation of craniofacial growth in a patient with Kabuki makeup syndrome: a case report. Eur J Orthod. 2001;23(2):205-13. [ Links ]
8. Matsumoto N, Niikawa N. Kabuki make-up syndrome: a review. Am J Med Genet C Semin Med Genet. 2003;117C(1):57-65. [ Links ]
9. Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31(3):565-89. [ Links ]
10. Maas NM, Van de Putte T, Melotte C, Francis A, Schrander-Stumpel CT, Sanlaville D, et al. The C20orf133 gene is disrupted in a patient with Kabuki syndrome. J Med Genet. 2007;44(9):562-9. [ Links ]
11. Armstrong L, Aleck K, Aughton DJ, Baumann C, Braddock SR, Gillessen-Kaesbach G, et al. Further delineation of Kabuki syndrome in 48 well-defined new individuals. Am J Med Genet A. 2005;132A(3):265-72. [ Links ]
12. Kawame H, Hannibal MC, Hudgins L, Pagon RA. Phenotypic spectrum and management issues in Kabuki syndrome. J Pediatr. 1999;134(4):480-5. [ Links ]
13. Niikawa N. Kabuki make-up (Niikawa-Kuroki) syndrome. Ryoikibetsu Shokogun Shirizu. 2001(36):484-5. [ Links ]
14. Wilson GN. Thirteen cases of Niikawa-Kuroki syndrome: report and review with emphasis on medical complications and preventive management. Am J Med Genet. 1998;79(2):112-20. [ Links ]
15. Digilio MC, Marino B, Toscano A, Giannotti A, Dallapiccola B. Congenital heart defects in Kabuki syndrome. Am J Med Genet. 2001;100(4):269-74. [ Links ]
16. Iida T, Park S, Kato K, Kitano I. Cleft palate in Kabuki syndrome: a report of six cases. Cleft Palate Craniofac J. 2006;43(6):756-61. [ Links ]
17. White SM, Thompson EM, Kidd A, Savarirayan R, Turner A, Amor D, et al. Growth, behavior, and clinical findings in 27 patients with Kabuki (Niikawa-Kuroki) syndrome. Am J Med Genet A. 2004;127A(2):118-27. [ Links ]
18. Schrander-Stumpel C, Meinecke P, Wilson G, Gillessen-Kaesbach G, Tinschert S, König R, et al. The Kabuki (Niikawa-Kuroki) syndrome: further delineation of the phenotype in 29 non- Japanese patients. Eur J Pediatr. 1994;153(6):438-45. [ Links ]
19. Hughes HE, Davies SJ. Coarctation of the aorta in Kabuki syndrome. Arch Dis Child. 1994;70(6):512-4. [ Links ]
20. Galan-Gomez E, Cardesa-García JJ, Campo-Sampedro FM, Salamanca-Maesso C, Martínez-Frías ML, Frías JL. Kabuki make-up (Niikawa-Kuroki) syndrome in five Spanish children. Am J Med Genet. 1995;59(3):276-82. [ Links ]
21. Ilyina H, Lurie I, Naumtchik I, Amoashy D, Stephanenko G, Fedotov V, et al. Kabuki make-up (Niikawa-Kuroki) syndrome in the Byelorussian register of congenital malformations: ten new observations. Am J Med Genet. 1995;56(2):127-31. [ Links ]
22. Mhanni AA, Cross HG, Chudley AE. Kabuki syndrome: description of dental findings in 8 patients. Clin Genet. 1999;56(2):154-7. [ Links ]
23. Kluijt I, van Dorp DB, Kwee ML, Toutain A, Keppler-Noreuil K, Warburg M, et al. Kabuki syndrome - report of six cases and review of the literature with emphasis on ocular features. Ophthalmic Genet. 2000;21(1):51-61. [ Links ]
24. McGaughran J, Aftimos S, Jefferies C, Winship I. Clinical phenotypes of nine cases of Kabuki syndrome from New Zealand. Clin Dysmorphol. 2001;10(4):257-62. [ Links ]
25. Shotelersuk V, Punyashthiti R, Srivuthana S, Wacharasindhu S. Kabuki syndrome: report of six Thai children and further phenotypic and genetic delineation. Am J Med Genet. 2002;110(4):384-90. [ Links ]
26. Ben-Omran T, Teebi AS. Structural central nervous system (CNS) anomalies in Kabuki syndrome. Am J Med Genet A. 2005;137(1):100-3. [ Links ]
27. Engelen JJ, Loneus WH, Vaes-Peeters G, Schrander-Stumpel CT. Kabuki syndrome is not caused by an 8p duplication: a cytogenetic study in 20 patients. Am J Med Genet A. 2005;132A(3):276-7. [ Links ]
28. Schoumans J, Nordgren A, Ruivenkamp C, Brøndum-Nielsen K, Teh BT, Annéren G, et al. Genome-wide screening using array-CGH does not reveal microdeletions/microduplications in children with Kabuki syndrome. Eur J Hum Genet. 2005;13(2):260-3. [ Links ]
29. Sanlaville D, Genevieve D, Bernardin C, Amiel J, Baumann C, de Blois MC, et al. Failure to detect an 8p22-8p23.1 duplication in patients with Kabuki (Niikawa-Kuroki) syndrome. Eur J Hum Genet. 2005;13(5):690-3. [ Links ]
30. Turner C, Lachlan K, Amerasinghe N, Hodgkins P, Maloney V, Barber J, et al. Kabuki syndrome: new ocular findings but no evidence of 8p22-p23.1 duplications in a clinically defined cohort. Eur J Hum Genet. 2005;13(6):716-20. [ Links ]
31. Kimberley KW, Morris CA, Hobart HH. BAC-FISH refutes report of an 8p22-8p23.1 inversion or duplication in 8 patients with Kabuki syndrome. BMC Med Genet. 2006;7:46. [ Links ]
32. dos Santos BM, Ribeiro RR, Stuani AS, de Paula e Silva FW, de Queiroz AM. Kabuki make-up (Niikawa-Kuroki) syndrome: dental and craniofacial findings in a Brazilian child. Braz Dent J. 2006;17(3):249-54. [ Links ]
33. Gabrieli AP, Rovaris F, Elaine L, Borges L, Michelin M, Lovatto L. Síndrome da maquiagem de kabuki. Acta Ortop Bras. 2002;10(3):57-61. [ Links ]
34. Morales E, Chiong C, Dyce E. Síndrome Kabuki. Presentación de dos casos. Revista Cubana de Genética Comunitaria. 2008;2(2):73-6. [ Links ]
35. Rocha C, Peixoto I, Fernandes P, Torres C, de Queiroz A. Dental findings in Kabuki make-up syndrome: a case report. Spec Care Dentist. 2008;28(2):53-7. [ Links ]
36. Silva E, Freitas E, Costa S, Duarte A. Down syndrome associated with clinical manifestations of Kabuki syndrome: report of a case. J Pediatr (Rio J). 1999;75(5):367-9. [ Links ]
37. Valdez-Geraldo C, García-Arias S, Valdez-Geraldo B, García-Torres O. Kabuki make-up syndrome. A case report with cognitive failure and haematologic features. Rev Neurol. 2009;48(5):278-9. [ Links ]
38. Teixeira C, Silva C, Honjo R, Bertola D, Albano L, Kim C. Dental evaluation of Kabuki syndrome patients. Cleft Palate Craniofac J. 2009;46(6):668-73. [ Links ]
39. Teixeira V, Neves M, de Castro R. Latex allergy in a patient with Kabuki syndrome. Case report. Rev Bras Anestesiol. 2010;60(5):544-50. [ Links ]
40. Guzmán-Acurio A, Tumbaco R, Jaramillo L. Sìndrome de Kabuki. Rev Ecuat Neurol. 2005;12:1-2. [ Links ]
41. Brito M, Nunes A. Intervenção Fonoaudiológica Na Síndrome De Kabuki: Relato De Caso. Rev CEFAC. 2010;12(4):693-9. [ Links ]
42. Cassidy SB, Allanson JE. Management of genetic syndromes. 3ª ed. Hoboken, N.J: Wiley-Blackwell; 2010. pp.962. [ Links ]
43. Kuniba H, Sato D, Yoshiura K, Ohashi H, Kurosawa K, Miyake N, et al. No mutation in RAS-MAPK pathway genes in 30 patients with Kabuki syndrome. Am J Med Genet A. 2008;146(14):1893-6. [ Links ]
44. Philip N, Devriendt K, Clayton-Smith J. Management of Kabuki Syndrome: A Clinical Guideline. 2010:1-23. [ Links ]
45. Mervis CB, Becerra AM, Rowe ML, Hersh JH, Morris CA. Intellectual abilities and adaptive behavior of children and adolescents with Kabuki syndrome: a preliminary study. Am J Med Genet A. 2005;132(3):248-55. [ Links ]
46. Vaux KK, Jones KL, Jones MC, Schelley S, Hudgins L. Developmental outcome in Kabuki syndrome. Am J Med Genet A. 2005;132(3):263-4. [ Links ]
47. Barozzi S, Di Berardino F, Atzeri F, Filipponi E, Cerutti M, Selicorni A, et al. Audiological and vestibular findings in the Kabuki syndrome. Am J Med Genet A. 2009;149(2):171-6. [ Links ]
48. Tekin M, Fitoz S, Arici S, Cetinkaya E, Incesulu A. Niikawa-Kuroki (Kabuki) syndrome with congenital sensorineural deafness: evidence for a wide spectrum of inner ear abnormalities. Int J Pediatr Otorhinolaryngol. 2006;70(5):885-9. [ Links ]
49. Moral S, Zuccarino F, Loma-Osorio P. Double aortic arch: an unreported anomaly with Kabuki syndrome. Pediatr Cardiol. 2009;30(1):82-4. [ Links ]
50. Dyamenahalli U, Abraham B, Fontenot E, Prasad V, Imamura M. Pathologic aneurysmal dilation of the ascending aorta and dilation of the main pulmonary artery in patients with Kabuki syndrome: valve-sparing aortic root replacement. Congenit Heart Dis. 2007;2(6):424-8. [ Links ]
51. Rosti RO, Kayserili H. Kabuki make-up syndrome with unilateral renal agenesis. Turk J Pediatr. 2009;51(3):298-300. [ Links ]
52. Iwama Y, Sugiyama S, Kaiga K, Eguchi M, Furukawa T. Kabuki makeup syndrome associated with megaureter. Acta Paediatr Jpn. 1987;29(1):182-5. [ Links ]
53. Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790-3. [ Links ]
54. Micale L, Augello B, Fusco C, Selicorni A, Loviglio MN, Silengo MC, et al. Mutation Spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J Rare Dis. 2011;6(1):38. [ Links ]
55. Banka S, Veeramachaneni R, Reardon W, Howard E, Bunstone S, Ragge N, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20(4):381-8.
56. Paulussen AD, Stegmann AP, Blok MJ, Tserpelis D, Posma-Velter C, Detisch Y, et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat. 2011;32(2):2018-25. [ Links ]
57. Aguilera S, Botella MP, Ocio I. Síndrome de Kabuki en el diagnóstico diferencial de hipotonía neonatal. An Pediatr. 2009;70(1):91-3. [ Links ]
58. Shah M, Bogucki B, Mavers M, deMello DE, Knutsen A. Cardiac conduction abnormalities and congenital immunodeficiency in a child with Kabuki syndrome: case report. BMC Med Genet. 2005;6:28. [ Links ]
59. Hou JW. Variable expressivity in a family with Kabuki make-up (Niikawa-Kuroki) syndrome. Chang Gung Med J. 2004;27(4):307-11. [ Links ]
60. Milunsky JM, Maher TA, Zhao G, Huang XL, Wang Z, Zou Y. A re-examination of the chromosome 8p22-8p23.1 region in Kabuki syndrome. Clin Genet. 2008;73(5):502-3. [ Links ]
61. Milunsky JM, Huang XL. Unmasking Kabuki syndrome: chromosome 8p22-8p23.1 duplication revealed by comparative genomic hybridization and BAC-FISH. Clin Genet. 2003;64(6):509-16. [ Links ]
62. Shieh JT, Hudgins L, Cherry AM, Shen Z, Hoyme HE. Triplication of 8p22-8p23 in a patient with features similar to Kabuki syndrome. Am J Med Genet A. 2006;140(2):170-3. [ Links ]
63. Miyake N, Harada N, Shimokawa O, Ohashi H, Kurosawa K, Matsumoto T, et al. On the reported 8p22-p23.1 duplication in Kabuki make-up syndrome (KMS) and its absence in patients with typical KMS. Am J Med Genet A. 2004;128(2):170-2. [ Links ]
64. Cusco I, del Campo M, Vilardell M, González E, Gener B, Galán E, et al. Array-CGH in patients with Kabuki-like phenotype: identification of two patients with complex rearrangements including 2q37 deletions and no other recurrent aberration. BMC Med Genet. 2008;9:27. [ Links ]
65. Kuniba H, Yoshiura K, Kondoh T, Ohashi H, Kurosawa K, Tonoki H, et al. Molecular karyotyping in 17 patients and mutation screening in 41 patients with Kabuki syndrome. J Hum Genet. 2009;54(5):304-9. [ Links ]
66. van Haelst MM, Brooks AS, Hoogeboom J, Wessels MW, Tibboel D, de Jongste JC, et al. Unexpected life-threatening complications in Kabuki syndrome. Am J Med Genet. 2000;94(2):170-3. [ Links ]
67. Chen CP, Lin SP, Tsai FJ, Chern SR, Wang W. Kabuki syndrome in a girl with mosaic 45,X/47,XXX and aortic coarctation. Fertil Steril. 2008;89(6):1826-7. [ Links ]
68. Chrzanowska KH, et al. Kabuki (Niikawa-Kuroki) syndrome associated with immunodeficiency. Clin Genet. 1998;53(4):308-12. [ Links ]
69. Ciprero KL, et al. Symptomatic Chiari I malformation in Kabuki syndrome. Am J Med Genet A. 2005;132(3):273-5. [ Links ]
70. David, G., et al. A case of Kabuki (Niikawa-Kuroki) syndrome associated with manifestations resembling C-trigonocephaly syndrome. Am J Med Genet A. 2004;130(4):389-92. [ Links ]
71. Ohdo S, et al. Kabuki make-up syndrome (Niikawa-Kuroki syndrome) associated with congenital heart disease. J Med Genet. 1985;22(2):126-7. [ Links ]
72. Shahdadpuri R, et al. A novel constellation of cardiac findings for Kabuki syndrome: hypoplastic left heart syndrome and partial anomalous pulmonary venous drainage. Pediatr Cardiol. 2008;29(4):820-2. [ Links ]
