










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.25 no.2 Bicaramanga May/Aug. 2012
Amiloidosis primaria asociada a mielofibrosis
idiopática crónica. Una relación inusual
Rafael Pila Pérez*
Pedro Rosales Torres**
Rafael Pila Peláez***
Víctor A. Holguín Prieto****
Lázaro Ramírez Lana*****
*MD Internista de II grado. Profesor Titular y Consultante. Servicio de Medicina Interna. Hospital Universitario Manuel Ascunce Domenech. Camagüey. Cuba.
**MD Patólogo de I grado. Profesor Instructor. Servicio de Medicina Interna. Hospital Universitario Manuel Ascunce Domenech. Camagüey. Cuba.
***MD Internista de II grado. Profesor Auxiliar. Servicio de Medicina Interna. Hospital Universitario Manuel Ascunce Domenech. Camagüey. Cuba.
****MD Internista de I grado. Servicio de Medicina Interna. Hospital Universitario Manuel Ascunce Domenech. Camagüey. Cuba.
*****MD Cardiólogo de II grado. Profesor Auxiliar. Servicio de Medicina Interna. Hospital Universitario Manuel Ascunce Domenech. Camagüey. Cuba.
Correspondencia: Dr. Rafael Pila Pérez. Dirección: Calle General Gómez # 452. Camagüey. Cuba. Código postal: 70100. e-mail: vadolfo@finlay.cmw.sld.cu.
Artículo recibido el 3 de mayo de 2012 y aceptado para publicación el 20 de julio de 2012.
RESUMEN
Introducción: las amiloidosis son entidades caracterizadas por la acumulación extracelular de material fibrilar en cantidad suficiente para comprometer las funciones vitales del órgano afectado. El componente fibrilar mayor determina el comportamiento y la clasificación. Presentación de caso: se presenta el caso de un paciente con amiloidosis primaria asociada a una mielofibrosis idiopática crónica con hematopoyesis extramedular esplénica. Se trata de un paciente masculino de 61 años que ingresa con el diagnóstico de una miocardiopatía restrictiva infiltrativa, alteraciones hematológicas, hepatoesplenomegalia y polineuropatía sensitivo-motora. El estudio mediante biopsia medular confirma el diagnóstico de amiloidosis, mientras que la citología por aspiración con aguja fina del bazo demuestra hematopoyesis extramedular y junto a los estudios hematológicos se concluye como una mielofibrosis idiopática crónica. Conclusiones: en la institución, los estudios de imagen e histopatológicos son fundamentales para confirmar la presencia de la amiloidosis. La asociación de amiloidosis primaria y mielofibrosis idiopática crónica fue casual, constituyendo el primer reporte en la literatura. (MÉD.UIS. 2012;25(2):137-44)
Palabras Clave: Amiloidosis. Mielofibrosis. Hematopoyesis Extramedular.
ABSTRACT
Primary amyloidosis associated to chronic idiopathic myelofibrosis. An unusual relationship
Introduction: amyloidosis is the term used for diseases caused by the extracellular deposition of protein fibrils in tissues and organs. These diseases are defined and classified by the biochemical nature of the protein in the fibril deposits. Clinical case: it is presented a clinical case of a patient with the diagnosis of primary amyloidosis and chronic idiopathic myelofibrosis with extramedullary hematopoiesis in the spleen. A 61-year male patient is admitted due to presenting infiltrative restrictive cardiomyopathy, hematologic alterations, hepatosplenomegalia and sensorimotor neuropathy. Amyloidosis is diagnosed through bone marrow biopsy, while fine-needle aspiration cytology of the spleen demonstrates extramedullary hematopoiesis, and along with hematologic studies, it is concluded as a chronic idiopathic myelofibrosis. Conclusions: in Universitary Hospital, imaging and histological studies are important to confirm the diagnosis of amyloidosis. The causality between primary amyloidosis and chronic idiopathic myelofibrosis there has not been reported in the literature. (MÉD.UIS. 2012;25(2):137-44)
Keywords: Primary Amyloidosis. Myelofibrosis. Extramedullary Hematopoiesis.
INTRODUCCIÓN
El término amiloidosis engloba un grupo de enfermedades de diversos orígenes, caracterizado por la acumulación de material ultraestructuralmente fibrilar en varios tejidos y en cantidad suficiente para comprometer las funciones vitales del órgano1. Los modernos análisis químicos han demostrado que con las técnicas convencionales de tinción, todos los amiloides aparecen homogéneos y eosinofílicos, se unen a la tinción rojo Congo y bajo la luz polarizada emiten una fluorescencia "verde manzana" birrefringente2.
La proteína amiloide es una proteína fibrilar, rígida, con estructura molecular terciaria en disposición ß-plegada, resistente a la digestión proteolítica y con agregados de fibras de 7,5-10 nm de ancho2,3. El componente P amiloide es una glucoproteína plasmática encontrada en todos los tipos de amiloide pero sin un papel patogénico conocido1,2,4. El catabolismo o proteólisis de las fibras amiloides es un factor importante en la patogénesis pero es pobremente conocido3. Los tipos de amiloides no pueden ser diferenciados por la distribución en los diferentes órganos ni por la microscopía electrónica2. El componente fibrilar mayor determina el comportamiento y la clasificación clínica. En el caso de la amiloidosis primaria, también llamada amiloidosis por cadenas ligeras de inmunoglobulinas se trata del amiloide AL (donde la A proviene de Amiloidosis y la L proviene de cadenas Ligeras) cuyo componente fibrilar resulta del depósito de cadenas ligeras de inmunoglobulinas, kappa (k) y lambda (λ), con predominio de las cadenas λ1,2.
Los pacientes con amiloidosis AL pueden tener una síntesis aberrante de novo o un proceso anormal proteolítico de las cadenas ligeras2,4. Raramente las fibras son cadenas monoclonales pesadas (amiloidosis AH, donde A corresponde a amiloidosis y H a cadenas pesadas del inglés heavy).
Las cadenas L y H de inmunoglobulinas solo son dos de las 20 diferentes proteínas fibrilares descritas en la amiloidosis humana con cuadros clínicos variables; señalándose a la amiloidosis AL relacionada con el mieloma múltiple y la gammapatía monoclonal de significado incierto. Por otro lado, El precursor proteico en la amiloidosis secundaria o amiloidosis A se asocia a varios procesos inflamatorios crónicos como: artritis reumatoide, espondilitis anquilosante, enfermedad de Still y Síndrome de Behçet; a infecciones sistémicas o locales crónicas, entre las que se encuentran lepra, osteomielitis, tuberculosis y bronquiectasia crónica; y ocasionalmente a neoplasias tales como enfermedad de Hodgkin, carcinoma de células renales, carcinoma gastrointestinal y carcinoma de pulmón1-6.
La metaplasia mieloide agnogénica, actualmente denominada Mielofibrosis Idiopática Crónica (MIC), es una enfermedad rara con una incidencia anual de 0,5-1,5 casos por 100 000 habitantes en los Estados Unidos, siendo más frecuente en personas de raza blanca, hombres y mujeres, la cual sufren por igual y la edad media del diagnóstico es de 65 años7. Algunos autores señalan que el elemento fundamental de la mielofibrosis es la hematopoyesis extramedular, la cual es invariablemente observada en bazo, hígado y ganglios linfáticos, pero puede observarse en otros sitios7,8. El 10-30% de los pacientes con MIC se diagnostican en fase asintomática ya sea por el hallazgo de un hemograma anormal o de una esplenomegalia8.
Ha motivado la presentación de este caso la rara asociación en un paciente de dos entidades hematológicas: una amiloidosis primaria y una MIC con hematopoyesis extramedular esplénica, el cual constituye el primer reporte en la literatura.
PRESENTACIÓN DE CASO
Se trata de un paciente masculino, de raza blanca, de 61 años de edad, con antecedentes de hipertensión arterial primaria hace 15 años, tratada con anticálcicos dihidropiridínicos y tiazidas, además de esplenomegalia de un año de evolución sin estudio sistemático. Hace tres meses comenzó a presentar astenia, fatiga, mareo, hipotensión y edemas en ambos miembros inferiores, síntomas asociados a ictericia de aparición gradual por lo que es hospitalizado con el diagnóstico de anemia hemolítica autoinmune por prueba de Coombs positiva. Al no encontrarse relación etiológica, se inicia tratamiento con glucocorticoides e inmunosupresores como azatioprina debido a la severidad de la hemólisis con cifras de Hb de 25-30 g/L, con deterioro del estado hemodinámico. Después de dos meses se le da el alta hospitalaria con seguimiento en consulta externa de hematología debido a la buena respuesta terapéutica. Siete días después comienza a presentar disnea a los moderados esfuerzos de empeoramiento progresivo, edemas blandos en miembros inferiores, hipotensión ortostática, palidez cutánea y fiebre. Es asistido en consulta de cardiología y en el estudio ecocardiográfico se observa una miocardiopatía con patrón restrictivo infiltrativo granular del miocardio. Debido a los episodios severos de hipotensión ortostática se suspenden los antihipertensivos.
Durante su estancia en la sala de Cardiología refiere dolores fulgurantes intensos, parestesias y contracturas de los dedos de las manos y de los pies con sensación de "alfilerazos" en la planta de los pies, lo cual se interpreta como una polineuropatía distal sensitivo-motora, iniciándose carbamazepina como tratamiento, con respuesta favorable. El paciente es trasladado a la sala de Medicina Interna para completar su estudio.
EXAMEN FÍSICO (EN EL MOMENTO DEL INGRESO):
Marcada afección del estado general, palidez mucocutánea, edema maleolar bilateral y temperatura de 37,8 ºC. Sistema cardiorrespiratorio: frecuencia respiratoria de 24 rpm, murmullo vesicular disminuido en ambas bases, no estertores, ruidos cardíacos arrítmicos por fibrilación auricular paroxística, no se auscultaron soplos, tensión arterial de 90/60 mmHg, frecuencia cardíaca central de 108 lpm. Abdomen: blando, globuloso, depresible, no doloroso a la palpación, hepatomegalia de 15 mm, de superficie lisa, ligeramente dolorosa a la palpación profunda. Sistema hemolinfopoyético: no se encontraron adenopatías, esplenomegalia estadio III de Boyd de 165 mm; bazo de consistencia dura, irregular, no doloroso. Sistema nervioso: consciente, sin signos meníngeos, con hallazgos compatibles con déficit sensitivo simétrico con distribución en guante y calcetín, se verifican signos de disfunción de motoneurona inferior. El resto del examen físico, incluyendo examen urológico y oftalmológico fue normal.
DATOS ANALÍTICOS (EN EL MOMENTO DEL INGRSO):
Estudio de lámina periférica: leucoeritoblastosis, granulocitos inmaduros escasos, eritrocitos nucleados, marcada anisopoiquilocitosis, glóbulos rojos en forma de lágrima (dacriocitos de Bessi), así como plaquetas grandes, atípicas y algunos megacariocitos fragmentados; inmunoelectroforesis en suero: disminución global de las inmunoglobulinas sin observarse paraproteína monoclonal; prueba de Sia: negativa; inmunoelectroforesis en orina (incluyendo técnica de concentración por ultrafiltración): ausencia de proteinuria de Bence- Jones, inmunoglobulinas o fragmentos de la misma. Proteinuria de 24 h: 3,1 g. VDRL, VIH 1 y 2, Ags HVB, Ac HVC: no reactivos; prueba de Mantoux con 5 UT (PPD): 2 mm, factor reumatoideo, ANA, crioglobulinas, niveles de complemento sérico: todos normales. Prueba de Coombs directa: positiva (ver Tabla 1).
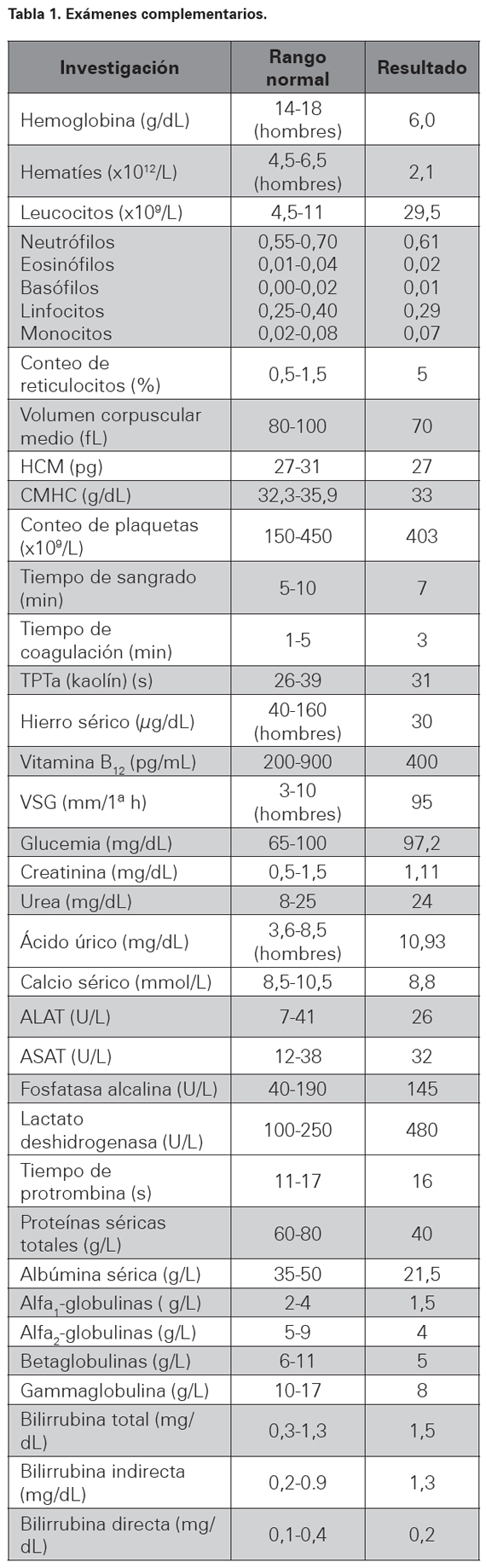
HCM: Hemoglobina corpuscular media, CMHC: Concentración media de hemoglobina corpuscular, TPTa: Tiempo parcial de tromboplastina activado, VSG: velocidad de sedimentación globular, ALAT: Alanino-aminotransferasa, ASAT: aspartatoaminotransferasa.
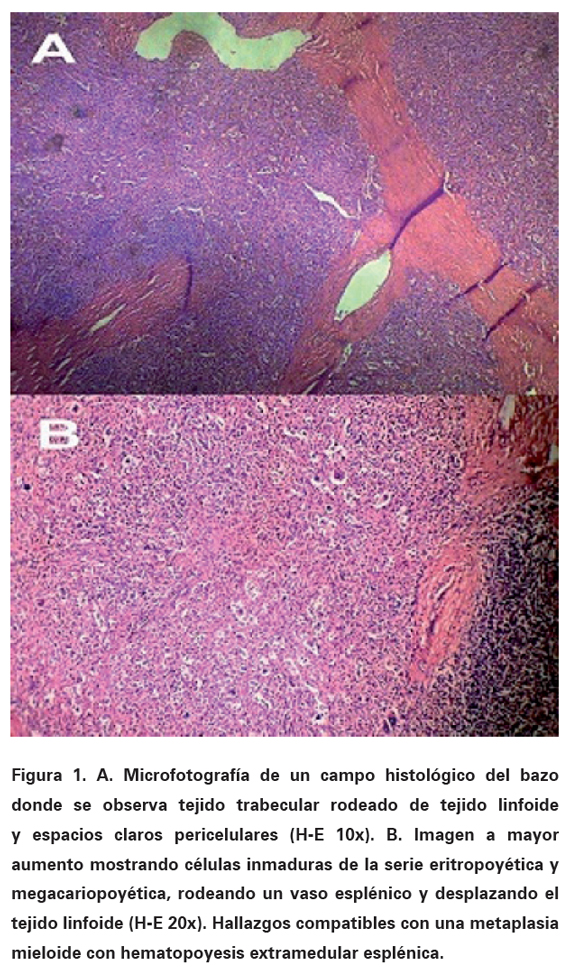
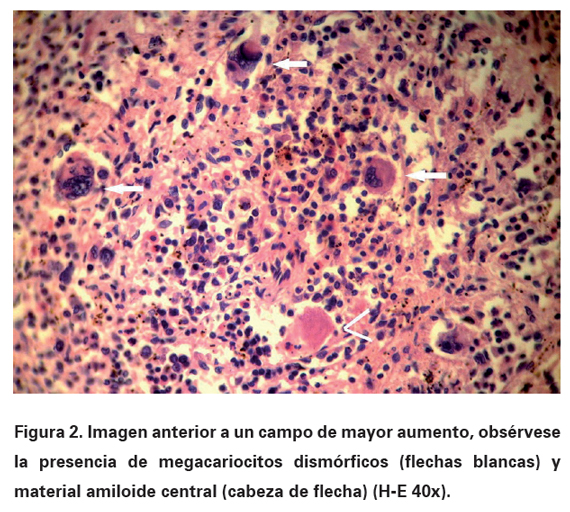
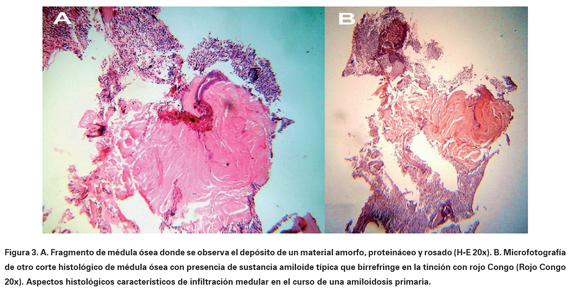
Electrocardiograma: bajo voltaje, fibrilación auricular, frecuencia cardiaca: 112 lpm, eje: +0o, contracciones ventriculares prematuras frecuentes, trastornos inespecíficos de la repolarización. Radiografía de tórax PA: índice cardiotorácico normal, borramiento bilateral del ángulo costofrénico. Ultrasonografía abdominal y de próstata: hepatomegalia no homogénea de 15 mm; esplenomegalia de 168 mm de superficie irregular, sin adenopatías; próstata con discreto aumento de volumen. Serie ósea (estudio radiológico), incluyendo cráneo, columna completa y pelvis ósea: normales. Ecocardiografía compatible con miocardiopatía restrictiva infiltrativa (miocardio de aspecto granular). Citología por aspiración con aguja fina del bazo (ver Figuras 1 y 2): metaplasia mieloide con presencia de precursores eritroides y numerosos megacariocitos; se observan algunos acúmulos aislados de material proteináceo eosinófilo en posible relación con depósito amiloide. Se indica aspirado medular, al cual se rehúsa el paciente. Se realiza biopsia de Médula Ósea (MO) (ver Figura 3) la cual demostró la existencia de infiltración amiloide por positividad de las tinciones de rojo Congo, ácido peryódico de Shiff y cristal violeta. Teniendo en cuenta los datos analíticos y los estudios histopatológicos se concluye el diagnóstico como una amiloidosis primaria o AL asociada a una Mielofibrosis Idiopática Crónica (MIC) con hematopoyesis extramedular esplénica. La amiloidosis secundaria fue excluida al no demostrarse alguna de las condiciones asociadas con la misma y ante la presencia de proteinuria con hipogammaglobulinemia.
Con este diagnóstico se indicó tratamiento quimioterápico para la amiloidosis AL de acuerdo con el régimen de la Clínica Mayo1-3,5 con melfalán a 0,15 mg/kg/día vía oral y prednisona a 0,8 mg/kg/día vía oral dividida en tres dosis durante siete días en ciclos de seis semanas, con estudios hematológicos frecuentes, hasta que se desarrolle leucopenia o trombocitopenia a la mitad del ciclo. Se adicionaron además alopurinol, testosterona, vitamina B6, ácido fólico y vitamina D. Para el manejo de la miocardiopatía se suspendieron los anticálcicos y se emplearon diuréticos. Durante su ingreso requirió en cuatro oportunidades transfusión de glóbulos rojos.
DISCUSIÓN
Los únicos estudios epidemiológicos sobre la prevalencia de la amiloidosis en el mundo se han practicado en los Estados Unidos, donde se reportan 1-5 casos por 100 000 habitantes anualmente, siendo los estudios de Minnesota los más destacados con un cálculo aproximado de 1 caso por 100 000 habitantes por año. Esta entidad afecta por igual a cualquier grupo social o étnico, siendo su frecuencia mayor en hombres que en mujeres en relación 2:1 con una edad media aproximada de 64 años9.
Típicamente la enfermedad afecta el corazón, el hígado, los riñones y el sistema nervioso periférico; más de la mitad de los pacientes mueren por afección cardíaca10, lo cual puede ocurrir por insuficiencia cardíaca diastólica o sistólica, arritmias o ambas11,12; no obstante, debe tenerse presente que otras entidades afectan el corazón con similares características infiltrativas como la sarcoidosis, la esclerosis sistémica progresiva, las metástasis cardíacas y la hemocromatosis13, traduciendo una miocardiopatía restrictiva1-3.
Las manifestaciones clínicas son muy variadas y dependen de los órganos afectados por el depósito amiloide, los principales signos incluyen: edema periférico, hepatomegalia, púrpura, hipotensión ortostática, neuropatía periférica, artritis, síndrome del tunel carpiano y macroglosia1-5. El edema periférico y la hipotensión arterial resultan de la insuficiencia cardíaca y del síndrome nefrótico3, lo cual se evidenció en el caso presentado.
Como se mencionó, el daño cardíaco puede manifestarse en forma de disfunción diastólica, insuficiencia cardíaca congestiva y arritmias, las cuales incluyen: bradicardia sinusal, bloqueo auriculoventricular, bloqueo de rama del haz de His, contracciones ventriculares prematuras y múltiples arritmias auriculares y ventriculares sin especificidad alguna por la amiloidosis cardíaca11. Es importante mencionar, tal y como se apreció en el paciente, que el uso de anticálcicos y digitálicos empeoran las manifestaciones cardiovasculares por su afinidad con el material amiloide; en el primer caso exacerba la insuficiencia por un mecanismo inotrópico negativo aumentado y en el segundo se aumenta la sensibilidad al fármaco desencadenando toxicidad12,14.
En el 20% de los casos ocurren depósitos de amiloide en los nervios periféricos causando neuropatía periférica de tipo sensitivo-motora, la cual es simétrica, traduce gran debilidad y muchas veces dolor, recordando a la polineuropatía diabética15; la neuropatía autonómica puede causar hipotensión ortostática y llevar al síncope3,5,13. Este tipo de manifestación fue observada en el paciente evaluado, quien mejoró con el empleo de la carbamazepina.
Los riñones son los sitios más frecuentes de depósito, siendo común la presentación en forma de un síndrome nefrótico, como en el paciente, algunos tienen rasgos de insuficiencia renal1,2. Es importante mencionar que una vez sospechada la presencia de amiloidosis, la interpretación del proteinograma es fundamental en el diagnóstico, pues la combinación de hipogammaglobulinemia y proteinuria sugiere la presencia de amiloidosis AL, en contraste, la amiloidosis renal secundaria es usualmente asociada con hipergammaglobulinemia debido a la inflamación persistente y a la producción de interleuquina 61.
No hubo motivos para pensar que existieran depósitos amiloides en otras localizaciones como la piel, el sistema endocrino y el sistema músculoesquelético, entre otros. La hepatoesplenomegalia es común y usualmente asintomáticas1. En el paciente, por las características de la hepatomegalia y el estudio de las enzimas hepáticas, nunca se tuvo en mente el daño hepático por amiloidosis, el cual puede observarse en el 25% de los casos9.
Es importante señalar que la no existencia de gammapatía monoclonal, proteinuria de Bence- Jones, ni de inflitrados plasmocitarios en la biopsia de MO, no descarta absolutamente un mieloma múltiple subyacente, entidad que frecuentemente se acompaña de amiloidosis como han referido muchos autores1-6.
Actualmente, la supervivencia media de los pacientes con amiloidosis es aproximadamente de 18 meses, por ello algunos autores14 consideran a la amiloidosis primaria una enfermedad semimaligna. La complicación cardíaca es extremadamente importante y ensombrece el pronóstico con solamente cuatro a seis meses de sobrevida después del comienzo de la insuficiencia cardíaca1,5,16. Sin embargo, los pacientes que solo presentan neuropatía periférica tienen una supervivencia prolongada de aproximadamente dos años, al igual que los que tienen una función renal normal y pocas células clonales en la biopsia de MO1,2,4,5.
El diagnóstico diferencial fundamental de esta enfermedad debe realizarse con los diferentes tipos de amiloidosis, lo cual en muchas ocasiones resulta difícil1-5.
Aunque hay cierta controversia en cuanto al mecanismo exacto de la fibrosis colágena en la MIC, hay unanimidad con respecto al papel central de los megacariocitos y las citocinas involucradas en la génesis de la fibrosis medular8. El 60% de los enfermos puede presentar palidez cutáneo-mucosa debido a múltiples mecanismos responsables de la anemia, incluyendo formas hemolíticas; signos hipermetabólicos como sudación, fiebre y pérdida de peso, y en el 20%, trastornos hemorrágicos con equimosis, entre otros7,17. Pueden ocurrir cuadros agudos con fiebre, dolores óseos e ictericia; la esplenomegalia se presenta en el 95-100% de los casos7,8, siendo la hepatomegalia discreta y bastante común (60-70%)8,18.
El cuadro hematológico apreciado en el leucograma y la lámina periférica, así como las características clínicas y la citología por aspiración con aguja fina del bazo demostraron que el paciente presentaba una MIC con hematopoyesis extramedular en el bazo asociados a una amiloidosis primaria, relación no reportada en la literatura hasta el momento, la cual se considera un hallazgo coincidente y no causal.
La biopsia de MO juega un papel importante para confirmar el diagnóstico de la MIC, para evaluar su severidad y para estimar la importancia de la hematopoyesis medular; el aspirado medular es de difícil realización debido a la mielofibrosis7,8. Si bien, mediante este estudio la MO demostró una infrecuente infiltración amiloide, el paciente presentó muchos de los criterios que definen a una MIC, tales como8:
- Cuadro leucoeritroblástico, anemia y glóbulos rojos en forma de lágrima llamados dacriocitos de Bessi.
- Esplenomegalia con evidencia de metaplasia mieloide.
- Ausencia de infecciones y otras enfermedades responsables de un diagnóstico alternativo.
Otro criterio diagnóstico es la ausencia de cromosoma de Filadelfia, el cual no se pudo realizar, constituyendo una limitante en este estudio.
La mediana de supervivencia de los pacientes con MIC es de unos 5 años7,8,17. Entre los factores pronósticos destacan la cifra de Hb (< 10 g/dL, el más importante), la sintomatología constitucional, la leucopenia o leucocitosis, la presencia de blastos en sangre periférica y las alteraciones citogenéticas7. En función de estos factores se reconocen formas de "bajo riesgo" y de "alto riesgo", con supervivencias significativamente diferentes (medianas de 8 y 2 años, respectivamente)8,17. El único tratamiento curativo de la MIC es el trasplante hematopoyético alogénico (convencional o con régimen de acondicionamiento de intensidad reducida).
Resultó muy llamativo durante la investigación clínica de este paciente descubrir la coexistencia de dos entidades hematológicas no relacionadas etiológicamente en ningún reporte médico; de allí que la presencia de una amiloidosis primaria junto a una MIC represente un hecho absolutamente casual y n o una asociación causal; más aún cuando se entiende que en la primera entidad la afección corresponde a una población de células plasmáticas de estirpe linfoide y en cambio la metaplasia mieloide constituye una de las expresiones del síndrome mieloproliferativo crónico de estirpe mieloide.
No existe un tratamiento satisfactorio para la amiloidosis1-5,9,15,19. Se han realizado estudios prospectivos con melfalán y prednisona utilizando dos tipos de régimen terapéuticos: el de la Clínica Mayo y el de la Universidad de Boston. En este caso se empleó el de la Clínica Mayo, con el régimen mencionado requiriéndose seis ciclos. Se ha utilizado también la colchicina sola o en combinación con melfalán y prednisona, sin embargo, la supervivencia media fue mayor en pacientes con melfalán, prednisona, y colchicina (18 meses) y en aquellos con melfalán y prednisona (17 meses) que en aquellos que recibieron la colchicina sola (8,5 meses)1-4,19. En la actualidad se emplean otros medicamentos tales como el bortezomib, un inhibidor de proteosomas aprobado para el manejo del mieloma múltiple, solo o unido a otros medicamentos20; también se han empleado carfilzomib, lenalidomida, talidomida y el idox, un análogo de antraciclina, el cual constituye la primera pequeña molécula con actividad in vivo capaz de solubilizar los depósitos amiloides de cadenas ligeras1-5. Aún permanece controversial la quimioterapia a altas dosis con trasplante de células madre en el manejo de la amiloidosis1, 2.
La hidroxiurea, el busulfán, la colchicina y el interferón alfa, entre otros, han demostrado gran utilidad en el tratamiento de la MIC7,8; no obstante al observar la respuesta de este enfermo con el régimen terapéutico para la amiloidosis, se decidió solo agregar hormonas masculinas, vitamina D, vitamina B6, ácido fólico y alopurinol, ya que el único tratamiento curativo para la MIC es el transplante alogénico de MO sobre todo en pacientes jóvenes7. La esplenectomía se reserva en general para infartos esplénicos, hipertensión portal, anemia progresiva bajo requerimiento de transfusiones o esplenomegalia sintomática que no ha respondido al tratamiento8.
Con este régimen terapéutico, el paciente mejoró ostensiblemente y recibió el alta hospitalaria con seguimiento en consulta externa de medicina interna y oncología. Después de ocho meses el paciente ha mantenido una evolución estable.
CONCLUSIONES
En el paciente no se evidenciaron antecedentes familiares de amiloidosis ni datos clínicos o analíticos a favor de un proceso crónico inflamatorio, infeccioso o tumoral en actividad, por lo que se descartaron la amiloidosis secundaria y la amiloidosis heredofamiliar; por todas estas razones se pensó que uno de los padecimientos analizados corresponde a una amiloidosis primaria con afección cardíaca, renal, de nervios periféricos y de la MO. Por otro lado, el estudio citológico de la esplenomegalia, junto al característico cuadro hematológico en el extendido de sangre periférica sugirió el diagnóstico de una MIC, si bien faltaron algunos elementos para su confirmación definitiva. La sintomatología expresada fue el resultado de un solapamiento de ambas entidades. Hasta el momento no existe reporte similar en la literatura, lo cual es una invitación a nuevos trabajos en este campo de la hematología. Debe resaltarse que aunque raras, son entidades que deben tenerse en cuenta en la clínica diaria por las implicaciones pronósticas para el paciente. La terapéutica fue dominada por la presencia de la amiloidosis, dado su pronóstico más sombrío, en particular cuando ya existe compromiso cardíaco.
REFERENCIAS BIBLIOGRÁFICAS
1. Wolf RE, Talavera F, Brent LH, McKenna R, Besa EC. Amyloidosis, Immunoglobulin-Related [monograph on the Internet]. NewYork: www.emedicine.com; 2010 [Last Updated: 2009 Feb 12]. Available from: http://emedicine.medscape.com/article/208839-overview [ Links ]
2. Gertz MA. Amyloidosis. In: Goldman L, Schafer AI, editors. Goldman's Cecil Medicine. 24th ed Philadelphia: Saunders Elsevier; 2012. p.1243-6. [ Links ]
3. Talavera F, Brent LH, Mechaber AJ, Diamond HS. Amyloidosis, Overview [monograph on the Internet]. New York: www.emedicine.com; 2010 [Last Updated: 2009 Jul 30]. Available from: http://emedicine.medscape.com/article/335414-overview [ Links ]
4. Bladé Creixenti J, San Miguel Izquierdo JF. Gammapatías monoclonales. En: Rozman C et al, editores. Medicina Interna Farreras-Rozman. 16a ed. Madrid: Ediciones Harcourt S.A; 2009, p. 1768-1777. [ Links ]
5. Buxbaum JN. The systemic amyloidoses. Curr Opin Rheumatol. 2004;16(1):67-75. [ Links ]
6. Nakamura T. Clinical strategies for amyloid A amyloidosis secondary to rheumatoid arthritis. Mod Rheumatol. 2008;18(2):109-18. [ Links ]
7. Tefferi A. Polycythemias, essential thrombocythemia, and primary myelofibrosis. In: Goldman L, Schafer AI, editors. Goldman's Cecil Medicine. 24th ed. Philadelphia: Saunders Elsevier; 2012, p.1090-8. [ Links ]
8. Seiter K, Talavera F, Guthrie TH, McKenna R, Besa EC. Agnogenic Myeloid Metaplasia With Myelofibrosis [monograph on the Internet]. New York: www.emedicine.com; 2009 [Last Updated: 2008 Oct 8]. Available from: http://emedicine.medscape.com/article/197954-overview [ Links ]
9. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45-59. [ Links ]
10. Hoyer C, Angermann CE, Knop S, Ertl G, Störk S. Cardiac amyloidosis. Med Klin (Munich). 2008;103(3):153-60. [ Links ]
11. Kristen AV, Meyer FJ, Perz JB, Schonland SO, Hundemer M, Hegenbart U, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 Suppl):S151-5. [ Links ]
12. Tan CD, Ratliff NB, Young JB, Rodriguez ER. Nonischemic Cardiomyopathies. In: Topol EJ. Textbook of Cardiovascular Medicine. 3rd Edition. Philadelphia: Lippincott Williams & Wilkins; 2007, p. 1407-27. [ Links ]
13. Lubitz SA, Goldbarg SH, Mehta D. Sudden cardiac death in infiltrative cardiomyopathies: sarcoidosis, scleroderma, amyloidosis, hemachromatosis. Prog Cardiovasc Dis. 2008;51(1):58-73. [ Links ]
14. Sack FU, Kristen A, Goldschmidt H, Schnabel PA, Dengler T, Koch A, et al. Treatment options for severe cardiac amyloidosis: heart transplantation combined with chemotherapy and stem cell transplantation for patients with AL-amyloidosis and heart and liver transplantation for patients with ATTR-amyloidosis. Eur J Cardiothorac Surg. 2008;33(2):257-62. [ Links ]
15. Seldin DC, Skinner M. Amyloidosis. In: Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J, editors. Harrison's Principles of Internal Medicine [CD-ROM]. 18th ed. New York: McGraw-Hill; 2012. [ Links ]
16. Bosch Genover X. Manifestaciones cardiovasculares de las enfermedades sistémicas. En: Rozman C et al, editores. Medicina Interna Farreras-Rozman [CD-ROM]. 14a ed. Madrid: Ediciones Harcourt S.A; 2000. [ Links ]
17. Dupriez B, Morel P, Demory JL, Lai JL, Plantier I, Bauters F. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88:1013-8. [ Links ]
18. Cervantes F, Barosi G, Demory JL, Reilly J, Guarnone R, Dupriez B, et al. Myelofibrosis with myeloid metaplasia in young individuals: disease characteristics, prognostic factors and identification of risk groups. Br J Haematol. 1998;102:684-90. [ Links ]
19. Kyle RA, Gertz MA, Greipp PR, Witziq TE, Lust JA, Lacy MQ, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202-7. [ Links ]
20. Jagannath S, Durie BGM, Wolf JL, Camacho ES, Irwin D, Lutzky J, et al. Long-term follow-up of patients treated with bortezomib alone and in combination with dexamethasone as frontline therapy for multiple myeloma. Blood. 2006;108:238. [ Links ]
