










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.26 no.2 Bicaramanga May/Aug. 2013
Caracterización clínica, estudios genéticos, y
manejo de la Mucopolisacaridosis tipo IV A
Jorge Luis Suárez-Guerrero*
Angie Katherine Bello Suárez**
María Carolina Vargas Santos***
Gustavo Adolfo Contreras-García****
*Estudiante VII semestre de Medicina. Grupo de investigación en Genética Humana. Director del grupo de formación en genética de SEIMED-UIS. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
**Estudiante XI semestre de Medicina. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
***Estudiante VII semestre de Medicina. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
****MD Genetista Médico. Docente en Genética Clínica. Grupo de investigación en Genética Humana. Universidad Industrial de Santander.
Departamento de Pediatría-Hospital Universitario de Santander. Bucaramanga. Santander. Colombia.
Correspondencia: Sr. Jorge Suárez. Calle 4 # 12-56 Nuevo Villabel. Floridablanca. Santander. Colombia. Teléfono: 3163300951. Correo electrónico: jorgesuarezg_@hotmail.com / jorgesuarezg@gmail.com. Teléfono: 3183970652.
Artículo recibido el 28 de mayo de 2013 y aceptado para publicación el 27 de julio de 2013
RESUMEN
Introducción: la Mucopolisacaridosis tipo IV A (OMIM #253000), es una enfermedad autosómica recesiva que pertenece al grupo de enfermedades de depósito lisosomal, esta fue descrita inicialmente por Luis Morquio, cuya etiología es la deficiencia de la enzima N-acetilgalactosamina- 6-sulfato sulfatasa, favoreciendo el depósito intracelular de queratán sulfato y condroitin-6-sulfato, llevando al espectro de manifestaciones clínicas que caracterizan este síndrome como son: baja talla, anormalidades vertebrales, opacidades corneales, inteligencia conservada, entre otras. Mediante radiografía de tórax y de extremidades inferiores se pueden observar las vértebras ovoides o en cuña y alteraciones en huesos largos, respectivamente. Objetivo: revisar sobre las generalidades de la MPS IV A, sus características clínicas, complicaciones, los estudios genéticos, la asesoría genética y su manejo preventivo. Conclusiones: las pruebas de laboratorio como el test de cloruro de cetilpiridinio o la albúmina ácida son esenciales para el diagnóstico. En cuanto al tratamiento hasta la fecha no existe una terapia de reemplazo enzimático por ello los cuidados son preventivos, y el manejo de estas personas debe ser interdisciplinario (medicina, nutrición, psicología, entre otros). (MÉD.UIS.2013;26(2):43-50)
Palabras Clave: Mucopolisacaridosis IV. Enfermedad de Morquio. Deficiencia de galactosamina-6-sulfato.
Mucopolysaccharidosis type IV A (Morquio Syndrome type A): clinical features, genetic
studies, preventive management of complications and genetic counseling.
ABSTRACT
Background: mucopolysaccharidosis IV A (OMIM #253000), belongs to the group of lysosomal storage diseases, this was first described by Luis Morquio, whose etiology is a deficiency of the enzyme N-acetylgalactosamine-6-sulfate sulfatase, favoring the deposit intracellular queratán sulfate and chondroitin-6-sulfate, leading to the spectrum of clinical manifestations that characterize this syndrome are short stature, vertebral abnormalities, corneal opacities, preserved intelligence, among others. X-ray can see the vertebrae ovoid or wedge, and alterations in long bones. Objetive: make a general overview of MPS IV A, its clinical features, complications, genetic testing, genetic counseling and preventive management. Conclusions: laboratory tests as test test cetylpyridinium chloride or acid albumin essential for diagnosis. As for treatment to date there is no enzyme replacement therapy are therefore preventive care, and management of these people should be interdisciplinary (medicine, nutrition, psychology, etc.). (MÉD.UIS.2013;26(2):43-50)
Keywords: Mucopolysaccharidosis IV. Morquio Disease. Galactosamine 6 Sulfatase Deficiency.
INTRODUCCIÓN
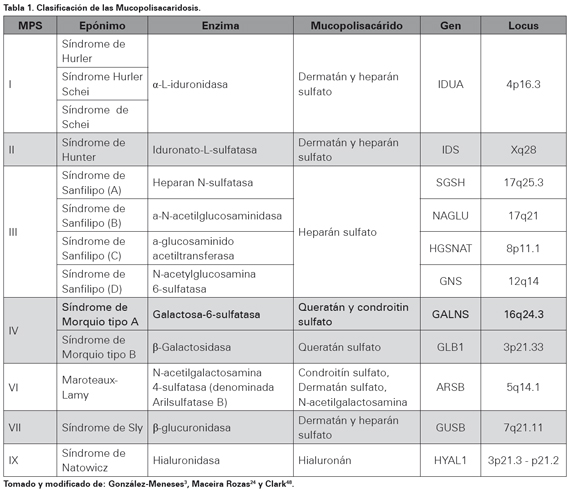
Se han denominado Mucopolisacaridosis (MPS) al conjunto de patologías de depósitos lisosomales, las que como grupo heterogéneo tienen en común la acumulación intracelular de sustancias intermedias del metabolismo de los mucopolisacaridos también denominados Glucosaminoglucanos (GAGs) se produce secundaria a una deficiencia enzimática lisosomal1-3. Actualmente se conocen siete tipos de MPS: tipo I o síndrome de Hurler, tipo II o Síndrome de Hunter, tipo III o síndrome de Sanfilipo, tipo IV o síndrome de Morquio, tipo VI o Marotaux-Lamy, tipo VII o síndrome de Sly y tipo IX o síndrome de Natowicz (Ver Tabla 1).
El objetivo del artículo es presentar una revisión sobre las generalidades de la MPS IV A, sus características clínicas, complicaciones, los estudios genéticos, la asesoría genética y su manejo preventivo.
MUCOPOLISACARIDOSIS TIPO IV A
Los primeros casos de MPS tipo IV fueron descritos por Luis Morquio, médico de origen uruguayo quien adelantó sus estudios de Pediatría en París y trabajó junto con Antoine Bernard-Jean Marfan, Jean-Martin Charcot, entre otros4,5. Después de su regreso a Uruguay, se dedicó a la docencia, fue cofundador de la Sociedad Uruguaya de Pediatría, y de la revista Archivo Latinoamericano de Pediatría4. Esto le permitió trabajar tanto en clínica como en academia, donde se dedicó a estudiar los desórdenes congénitos y adquiridos. En 1929 describió el caso de cuatro hermanos con distrofias esqueléticas clasificadas, en ese entonces, como distrofias óseas familiares; ese mismo año Brailsford, radiólogo inglés, describió un paciente de cuatro años de edad con la misma condición, nombrándole osteocondrodistrofia6. Morquio, especificó cambios radiológicos observables antes de la aparición de los signos y síntomas clínicos que suelen aparecer hacia los tres años de edad7. Con el tiempo esta enfermedad genética recibió el nombre de Síndrome de Morquio, Síndrome Morquio-Brailsford o MPS tipo IV4,8. Actualmente hay una distinción entre dos variantes de la misma MPS conocidas como tipo IV A y Tipo IV B.
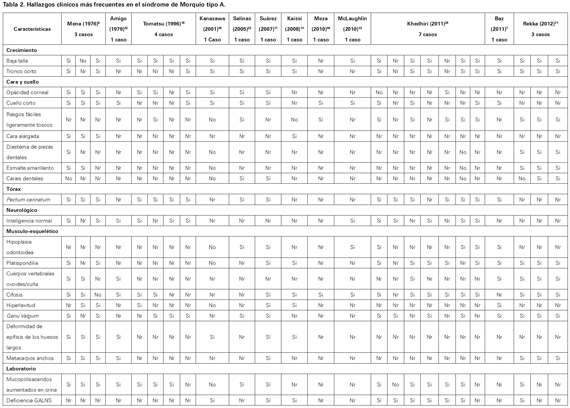
La MPS tipo IV A, MPS IV A o Síndrome de Morquio A (OMIM # 253 000) es una enfermedad autosómica recesiva donde existe afección del sistema osteoarticular y del tejido de sostén, en la cual la gravedad es dependiente de la actividad realizada por la enzima deficiente1,9-11. Las principales alteraciones en estas personas son la baja talla, tronco corto, pectus carinatum, cifoescoliosis, hiperlaxitud, inestabilidad de la columna cervical y vértebras en cuña; la inteligencia se conserva pero la deformidad del tronco puede comprimir la médula hasta generar debilidad progresiva y parálisis3,6,12-9.Es la forma más común, primeramente reconocida y mas severa.
Por su parte la MPS tipo IV B (OMIM # 256 540), enfermedad autosómica recesiva menos frecuente, se considera la más leve forma y se caracteriza por deficiencia de la enzima β-galactosidasa (el gen se ubica en 3p24.3) cuyo resultado es la acumulación de queratán sulfato, presentando manifestaciones clínicas similares a la MPS IV A3,20,21. Su incidencia estimada varía de 1/75 000 a 1/640 000 nacidos vivos20.
CARACTERIZACIÓN GENÉTICA
La MPS IV A se halla dentro de las MPS descritas inicialmente por Mc Kusick y colaboradores basadas en un orden numérico y según el tipo de glucosaminoglicano excretado en orina junto a las características clínicas y las que se han ido modificando con la identificación de nuevas enzimas en los últimos años; presenta un mecanismo de herencia de tipo autosómico recesivo con una frecuencia variable a nivel mundial de 1/75 000 a 1/200 0001,8,14,17,22. La MPS IV A es causada por la deficiencia de la enzima hidrolasa lisosomal, la N-Acetilgalactosamina-6-sulfato sulfatasa o GALN (por sus siglas en inglés), que tiene la función de hidrolizar el condroitín sulfato de la N-acetil-Dgalactosamina 6-sulfato y las unidades de queratán sulfato de la D-galactosa 6-sulfato23. El gen ubicado en 16q24.31,3,10,14,24 posee 2339 nucleótidos, contiene 14 exones, 13 intrones y su secuencia codifica un polipéptido de 522 aminoácidos1,9,25. Los estudios han reportado 16 polimorfismos y aproximadamente 187 mutaciones11,18,26-8, de las cuales tres mutaciones sin sentido son las más frecuentes: p.R386C, p.G301C, y p.I113F29. El depósito intracelular de dichos componentes, desencadena el daño permanente y progresivo particularmente en los fibroblastos y leucocitos, así como la excreción urinaria de queratán sulfato y condroitín-6-sulfato13,15,17,30, lo que permite confirmar el diagnóstico mediante pruebas enzimáticas y moleculares.
CARACTERIZACIÓN CLÍNICA
Generalmente no existen manifestaciones morfológicas en neonatos, porque estas entidades se manifiesta con alteraciones, de severidad variable y aparición progresiva principalmente entre el primer a tercer año de vida1,21, comprometiendo el desarrollo y función del sistema osteo-articular y de sostén. Los primeros signos por lo general son los trastornos de la marcha con posición anormal de piernas ,deformidad de la caja torácica y lento crecimiento, son los signos de sospecha para el médico. Lo anterior genera las alteraciones clínicas que se han descrito propiamente en este síndrome1,3,6,8,11,13-8,21, tales como la baja talla desproporcionada para la edad, perdiéndose la relación entre el tronco y las extremidades1,6,14,21; a nivel cráneo-facial las fascies toscas son menos marcadas que en las demás MPS, sin embargo presentan prognatismo, boca amplia, puente nasal plano, opacidades corneales, malformaciones y caries dentales, hipoacusia neurosensorial y de conducción, cuello corto, e hipoplasia odontoidea1,8,17,21,31.
Respecto a la región toraco-abdominal es importante tener presente la cifosis secundaria a malformaciones vertebrales descritas típicamente como vertebras en cuña o vertebras ovoides, la presencia de pectus carinatum, y tronco corto1,6,8,14,17,21. Las anteriores alteraciones pueden llevar a complicaciones cardíacas y pulmonares, como valvulopatías o neumopatías restrictivas.
Las extremidades inferiores suelen presentar genu valgum, pie plano, e hiperlaxitud articular a nivel de cadera, esto genera limitación en los movimientos, o incluso puede llevar a parálisis secundaria a la compresión medular torácica6-8,14. Las personas con el síndrome tienen un desarrollo psicomotor y coeficiente intelectual normal6,15-18,26,30,32,33.
La expectativa de vida depende de la gravedad de la enfermedad, presentando un margen que varía aproximadamente entre 20 y 70 años34,35. La muerte generalmente ocurre debido a una insuficiencia cardiorrespiratoria por deformidad esquelética torácica que compromete la vía aérea3,19 (Ver Tabla 2).
CARACTERIZACIÓN RADIOLÓGICA
En la radiografía de columna se puede observar la desviación de la columna secundaria a la presencia de cuerpos vertebrales anormales como en cuña u ovoides, de igual forma se pude encontrar hipoplasia de la apófisis odontoidea y platispondilia principalmente en T11, T12 y L513,14,17,26. En la radiografía de cadera y miembro inferior se pueden observar metacarpos cortos y anchos, coxa valga, así como cabezas femorales pequeñas o aplanamiento de las mismas aplanamiento de la cabeza femoral13,14,17,36.
PRUEBAS DE DETERMINACIÓN RECONOCIDAS
En general, el diagnóstico inicial de las MPS se realiza a partir de las observación de las manifestaciones clínicas en un individuo afectado generalmente de ocurrencia esporádica cuando no existen antecedentes de riesgo de recurrencia en las familias, seguidos de los hallazgos obtenidos de imágenes diagnósticas, en estudios imagenológicos de Rayos X, Ultrasonido, resonancia nuclear magnética y otros medios complementarios tales como los auditivos, oftalmológicos, entre otros. Su confirmación se hace mediante la determinación de niveles elevados de GAGs y la función enzimática (para diferenciar entre tipo A y B). Actualmente se están empleando técnicas para determinar las mutaciones específica que pueda presentar una persona tanto en MPS IV A como en MPS IV B3.
En la literatura se han planteado diferentes pruebas de tamizaje y diagnóstico empleadas en las MPS, sin embargo, algunas solo sirven para determinadas enfermedades, por ello a continuación se presentan las diferentes pruebas a realizar en MPS tipo IV según lo reportado en la literatura.
a. Detección de GAGs: mediante pruebas cuantitativas (test azul de dimetiletil, DMB por sus siglas dimethylethylene blue en inglés) o cualitativas (cromatografía en capa fina o electroforesis uni o bidireccional), se pueden medir los niveles de GAGs en sangre u orina, siendo esta última la más usada3,24. El más usado es el test de Cloruro de Cetilpiridinio, prueba turbidimétrica en donde la interacción entre los GAGs con sales de amonio genera un precipitado permitiendo así su cuantificación37. La prueba de la albúmina ácida consiste en la formación de turbidez lechosa en medio ácido ante la presencia de GAGs38. El test de azul de dimetiletil se emplea con muestras de orina, su mecanismo de acción se basa en la formación de complejos entre el dimetiletil y el GAGs generando una absorción espectofotométrica a 553 nm1,además de ser accesible y rápido, este test presenta una sensibilidad y especificidad superior al 90%1.
A pesar de lo anterior, este análisis no verifica de forma definitivamente el diagnóstico de MPS, solo determina una estimación de la concentración de GAGs, por lo que se requiere complementarlo con una valoración cualitativa, dada por la electroforesis de GAGs en muestra de orina sobre una placa de acetato de celulosa, la cual consiste en la separación en bandas de las macromoléculas según su carga eléctrica24,39; por su parte la cromatografía en capa fina permite la separación de dichos GAGs mediante su peso molecular y su afinidad por la fase móvil, permitiendo así su separación en bandas para el posterior análisis2,40. El método de espectrometría de masas en tándem es excelente como cribado en recién nacidos con alto riesgo para la enfermedad, obteniendo resultados del 100% de sensibilidad y especificidad en la determinación de marcadores de alteraciones lisosomales para la MPS tipo IV A24,41,42.
Otra técnica de reciente aparición para el diagnóstico de MPS es la prueba de ELISA24,43, en donde anticuerpos dirigidos contra epítopes específicos del queratán sulfato determinan específicamente su presencia en sangre y orina. Su valor varía con la edad y se ha visto una correlación entre la concentración de queratán sulfato y la severidad de la enfermedad, siendo significativa en muestra de orina (p < 0,001)43. Este método se utiliza cuando las pruebas de tamizaje inicial clásicas no son positivas aun cuando hay una fuerte sospecha clínica e imagenológica, ya que disminuye los falsos positivos y negativos de estas pruebas.
b. Determinación enzimática: concomitante con la detección de GAGs, se continúa con el diagnóstico enzimático confirmatorio, la determinación en papel de filtro permite medir mediante fluorescencia la actividad enzimática deficiente o con actividad disminuida en muestras de sangre seca24,44. Siempre debe confirmarse con determinación enzimática en leucocitos de sangre periférica24. Otras muestras como las vellosidades coriónicas o el líquido amniótico permite el análisis en etapa gestacional1.
c. Secuenciación de genes: si se desea conocer la alteración exacta que causa la enfermedad se puede establecer las mutaciones genéticas involucradas, y por ende una mejor visualización de polimorfismos y posible heterogeneidad de la enfermedad. El método más usado es la PCR1,45, pero existen otras técnicas las cuales usan la digestión enzimática para rastrear mutaciones puntuales, tales como el polimorfismo conformacional de simple cadena y sistema de amplificación refractaria de mutación específica1. La secuenciación puede hacerse de igual manera, como metodología para la búsqueda de la mutación en el gen de la MPS IV A24,45.
GUÍA DE MANEJO PREVENTIVO Y TERAPÉUTICO
La National MPS Society8, estableció guías de prevención y tratamiento. Dentro de las recomendaciones están las pruebas enzimáticas prenatales para el diagnóstico del síndrome en parejas que tienen antecedente de otro hijo afectado por la enfermedad. Por otro lado, en las personas con MPS IV, se debe tener presente el crecimiento, al presentar retardo del mismo, por ende el llevar a cabo curvas de crecimiento es de suma importancia. Respecto a la cifoescoliosis leve, esta puede disminuirse con correctores de postura; también en relación con la hipoplasia odontoidea, autores recomiendan corrección quirúrgica mediante la fusión de vertebras mejorando la estabilidad de C1 y C28,17 como en casos reportados22. De igual forma es importante tener presente el rápido agotamiento con la marcha o signos piramidales, indicadores de comprensión medular3. En las extremidades inferiores el genu valgum es común en MPS IV A, sin embargo no se recomienda ninguna cirugía correctiva hasta después de terminado el crecimiento. Es importante la terapia física en estos pacientes para mantener la fuerza muscular y movilidad adecuada para la vida diaria.
Como se ha mencionado, las personas afectadas presentan malformaciones torácicas que pueden causar restricciones respiratorias, por ello es importante que el especialista lleve a cabo pruebas para determinar el nivel de restricción existente, de igual forma las infecciones respiratorias y de oído medio suelen necesitar terapia antibiótica. En relación con la opacidad corneal, se recomienda usar gafas negras para evitar las molestias con la luz del día; y en la hipoacusia el manejo depende del origen de la misma, en caso de ser hipoacusia neurosensorial es necesario el implante coclear, mientras que para la hipoacusia de conducción se suelen insertar tubos de ventilación en el tímpano, favoreciendo presiones adecuadas. Las cardiopatías son complicaciones a tener presente, especialmente las valvulopatías por lo cual debe hacerse un ecocardiograma anualmente.
Si bien se trata de una enfermedad genética heredo metabólica con modelos de trasmisión hereditaria muy bien identificados y con una incidencia no tan alta en las poblaciones, la posibilidad de su prevención es posible no solo en aquellas familias conocidas en riesgo sino que hoy se habla de una prevención primaria a nivel comunitario. La prevención puede hacerse según las manifestaciones clínicas del paciente, o mediante la búsqueda activa de estas por edades en los grupos de riesgo, tal y como lo plantean Suárez y Zarante17, quienes recomiendan que en los controles entre los cero y un año de vida se haga énfasis en los signos de hidrocefalia, la opacidad corneal, la cifoescoliosis y la luxación de cadera; entre los uno a seis años tener presente las alteraciones óseas a nivel torácico, de cadera y columna, así como la afección cardiovascular. En los mayores de los seis años se debe tener presente la inestabilidad atlanto-axoidea con posible corrección quirúrgica, y el manejo por parte de ortopedia para las diferentes alteraciones esqueléticas propias del síndrome, aunque en términos generales desde el nacimiento el manejo debe ser interdisciplinar con la afección multiorgánica que padecen las personas con MPS IV A.
La inteligencia suele no afectarse en estas personas pero si debe evaluarse la afectación psicológica que deviene de asistir el paciente inicialmente sano a un grave y progresivo deterioro de su anatomía ósea y funciones conservando aun así su intelecto normal pero con capacidades cada vez más limitadas. De aquí la importancia del asesoramiento por parte de psicólogos, psiquiatras, y trabajadores sociales tanto a nivel del propio paciente como de los familiares y las comunidades, por lo que elaborar programas de ajustes psicológicos, redes de apoyo y mecanismos de inserción social y familiar son muy necesarios y valorados hoy día dentro de los preceptos de la medicina familiar.
A pesar que en la actualidad no hay un tratamiento curativo para esta enfermedad, recientemente han surgido algunas alternativas aún en estudio, como la terapia de reemplazo enzimático y terapia con células madres hematopoyéticas. Respecto a la terapia de reemplazo enzimático, aún se encuentra en fase experimental por ende su aplicación clínica aún no está disponible1,46-8, sin embargo, para algunas MPS el trasplante de médula ósea, el reemplazo enzimático, y el uso de células madre han sido considerados como opciones, ya que podría conseguir desacelerar la progresión o revertir algunos síntomas en determinados pacientes3,35, aunque estas técnicas no están libres de riesgos, limitaciones, y no está claro que pueda prevenir daños ulteriores a determinados órganos o tejidos. De todas formas actualmente el manejo multidisciplinario es paliativo y preventivo de las complicaciones tal y como lo sugieren las guías de manejo de la National MPS.
ASESORÍA GENÉTICA
La asesoría genética debe basarse en un diagnóstico correcto, que es resultado de la historia clínica, el examen físico y las pruebas complementarias (citogenéticas, moleculares, bioquímicas, etc). Además la asesoría está dirigida a explicar el riesgo de recurrencia teniendo en cuenta que es una enfermedad autosómica recesiva, por lo tanto se establecería en 25% para los padres de un individuo afectado. Debe tenerse en cuenta la enfermedad de novo que explica su aparición esporádica sin antecedentes familiares heredables. Factores sociales y culturales de algunas poblaciones con alto grado de endogamia, matrimonios no al azar, comunidades cerradas, religiones, han sido reconocidos como favorecedores de la unión de heterocigóticos sanos con descendencia afectada, con una probabilidad más alta que en una población general. Es muy útil un exitoso asesoramiento genético familiar como única alternativa para cortar las cadenas de trasmisión del gen afecto, a partir del reconocimiento de portadores sanos en los familiares, mucho más cuando en muchos países la población no dispone de los recursos que permiten un diagnóstico clínico de certeza y oportuno apoyado en complementarios de alta complejidad, por su escasa accesibilidad a los servicios de salud o el poco conocimiento de profesionales sobre aspectos de las enfermedades genéticas. En la medida de lo posible las patologías presentes deben ser tratadas por especialistas, mediante un trabajo interdisciplinar que debe estar dirigido por el pediatra, el médico internista o el genetista clínico, con el fin de conseguir los mejores resultados.
CONCLUSIONES
La MPS IV A o síndrome de Morquio es una enfermedad heredometabólica, de naturaleza autosómica recesiva que requiere cuidados especiales, pues debido a la expresión clínica fenotípica del defecto, estas personas suelen tener alteraciones óseas que comprometen diferentes órganos vitales, incluso puede ser causa de deceso a edades tempranas. Es por ello que se ha creado la guía de diagnóstico y manejo con el fin de mejorar la calidad y expectativa de vida de estas personas, siendo los profesionales de la salud los encargados de llevar a cabo dicha labor. El manejo interdisciplinario es de suma importancia para el diagnóstico, asesoramiento y orientación tanto al paciente como a los familiares.
REFERENCIAS BIBLIOGRAFÍCAS
1. Bouzidi H, Khedhiri S, Laradi S, Ferchichi S, Daudon M, Miled A. Mucopolysaccharidosis IVA (Morquio A syndrome): clinical, biological and therapeutic aspects. Ann Biol Clin. 2007;65(1):5-11. [ Links ]
2. Correa Garzón LN. Mucopolisacaridosis. Precop SCP. 2005;4(3):30-37. [ Links ]
3. González-Meneses López A, Barcia Ramírez A, Díaz-Rodríguez JL. Protocolo de actuación en las mucopolisacaridosis. Protoc diagn ter pediatr. 2010;1:24-36. [ Links ]
4. Chudley AE, Chakravorty C. Genetic landmarks through philately: Luis Morquio 1867-1935. Clin Genet. 2002;62(6):438-9. [ Links ]
5. Haas LF. Luis Morquio (1867-1935). J Neurol Neurosurg Psychiatry. 2002;72(6):787. [ Links ]
6. Mena M, Obando R. Sindrome de Morquio. Rev Chil Pediatr.1976;47(3):247-54. [ Links ]
7. Baz AB, Akalin S, Arik H, Ergün A. Proximal realignment surgery for unilateral chronic patella dislocation in Morquio syndrome: a case report. Acta Orthop Traumatol Turc. 2011;45(6):466-9. [ Links ]
8. National MPS Society. Guía Para Entender el Síndrome de Morquio. Durham: National MPS Society. p. 2-23. [ Links ]
9. Laradi S, Tukel T, Khediri S, Shabbeer J, Erazo M, Chkioua L. Mucopolysaccharidosis type IV: N-acetylgalactosamine-6-sulfatase mutations in Tunisian patients. Mol Genet Metab. 2006 Mar;87(3):213-8. [ Links ]
10. Baker E, Guo XH, Orsborn AM, Sutherland GR, Callen DF, Hopwood JJ, et al. The morquio A syndrome (mucopolysaccharidosis IVA) gene maps to 16q24.3. Am J Hum Genet. 1993;52(1):96-8. [ Links ]
11. Tomatsu S, Fukuda S, Cooper A, Wraith JE, Rezvi GM, Yamagashi A, et al. Mucopolysaccharidosis IVA: identification of a common missense mutation I113F in the N-Acetylgalactosamine-6-sulfate sulfatase gene. Am J Hum Genet. 1995;57(3):556-63. [ Links ]
12. Andreucci E, Aftimos S, Alcausin M, Haan E, Hunter W, Kannu P, et al. TRPV4 related skeletal dysplasias: a phenotypic spectrum highlighted byclinical, radiographic, and molecular studies in 21 new families. Orphanet J Rare Dis. 2011 Jun; 6:37. [ Links ]
13. Kaissi AA, LKlaushofer K, Grill F. Progressive acetabular dysplasia in a boy with mucopolysaccharoidosis type A (Morquio syndrome): a case report. Cases J. 2008;1:410-4. [ Links ]
14. López LS, Velásquez A, Abad M, Espinal D. Mucopolisacaridosis tipo IV como causa de talla baja patológica: reporte de un caso y revisión de la literatura. Rev CES Med. 2008;22(2):89-97 [ Links ]
15. Merritt HH, Rowland LP. Merritt's neurology. 10ª ed. Philadelphia: Lippincott Williams & Wilkins; 2000. p. 547. [ Links ]
16. Northover H, Cowie RA, Wraith JE. Mucopolysaccharidosis type IVA (Morquio syndrome): a clinical review. J Inherit Metab Dis. 1996;19(3):357-65. [ Links ]
17. Suárez Obando F, Zarante Montoya I. Aspectos clínicos y manejo integral del síndrome de Morquio. Univ Méd. 2007;48(2):166-74. [ Links ]
18. Tomatsu S, Fukuda S, Yamagishi A, Cooper A, Wraith JF, Hori T, et al. Mucopolysaccharidosis IVA: four new exonic mutations in patients with N-acetylgalactosamine-6-sulfate sulfatase deficiency. Am J Hum Genet. 1996;58(5):950-62. [ Links ]
19. Pelley CJ, Kwo J, Hess DR. Tracheomalacia in an adult with respiratory failure and Morquio syndrome. Respir Care. 2007;52(3):278-82. [ Links ]
20. Caciotti A, Garman SC, Rivera-Colón Y, Procopio E, Catarzi S, Ferri L, et al. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys Acta. 2011;1812(7):782-90. [ Links ]
21. Orphanet [sede Web]. Paris: Orphanet; 1997. Maire I, Froissart R. Mucopolysaccharidose de type IV A ou IV B ou Maladie de Morquio [3 páginas] [ Links ].
22. Salinas H, Preisler J, Astudillo J, Cerda S, Castillo S, Fernández F, et al. Síndrome de Morquio (Mucopolisacaridosis tipo IV) y embarazo. Rev Chil Obstet Ginecol. 2005;70(6):400-3. [ Links ]
23. AmiGo [base de datos en Internet]. USA: Gene Ontology Consortium; 1999 [acceso 2013]. N-acetylgalactosamine-6- sulfatase activity. Disponible en: http://amigo.geneontology.org/cgi-bin/amigo/term_details?term=GO:0043890#top. [ Links ]
24. Maceira Rozas MC, Atienza Merino G. Detección precoz de mucopolisacaridosis y oligosacaridosis en el período neonatal mediante cribado poblacional. Revisión sistemática. Madrid: Ministerio de Sanidad y Consumo; 2006. p. 21-124. [ Links ]
25. Tomatsu S, Fukuda S, Masue M, Sukegawa K, Fukao T, Yamagishi A, et al. Morquio disease: isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem Biophys Res Commun. 1991;181(2):677-83. [ Links ]
26. Khedhiri S,Chkioua L, Ferchichi S, Miled A, Laradi S. Polymorphisms in Tunisian patients with N-acetylgalactosamine- 6-sulfate sulfatase gene deficiency: implication in Morquio A disease. Diagn Pathol. 2011;6:11. [ Links ]
27. Sukegawa K, Nakamura H, Kato Z, Tomatsu S, Montaño AM, Fukao T, et al. Biochemical and structural analysis of missense mutations in N-acetylgalactosamine-6-sulfate sulfatase causing mucopolysaccharidosis IVA phenotypes. Hum Mol Genet. 2000;9(9):1283-90. [ Links ]
28. The Human Gene Mutation Database [sede Web]. Cardiff: Cardiff University; 2013. Galactosamine (N-acetyl)-6-sulphate sulphatase. disponible en: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GALNS. [ Links ]
29. Tomatsu S, Montaño AM, Nishioka T, Gutierrez MA, Peña OM, Tranda Firescu GG, et al. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum Mutat. 2005;26(6):500-12. [ Links ]
30. Clarke JTR. A clinical guide to inherited metabolic diseases. 3ª ed. Cambridge: Cambridge University Press; 2006. p.338. [ Links ]
31. Rekka P, Rathna PV, Jagadeesh S, Seshadri S. Mucopolysaccharidoses type IV A (Morquio syndrome): a case series of three siblings. J Indian Soc Pedod Prev Dent. 2012;30(1):66-9. [ Links ]
32. Amigo O, Burrows R, and Muzzo S. Mucopolisacaridos tipo IV. Síndrome de Morquio. Rev Chil Pediatr. 1979;50(3):61-4. [ Links ]
33. McLaughlin AM, Farooq M, Donnelly MB, Foley K. Anaesthetic considerations of adults with Morquio's syndrome - a case report. BMC Anesthesiol. 2010;10:2. [ Links ]
34. Pachajoa H, Rodríguez CA, Isaza C. Posible caso de síndrome de Morquio en la cerámica de la cultura tumaco-tolita. Rev Neurol. 2009;48(1):52. [ Links ]
35. Tomatsu S, Montaño AM, Oikawa H, Smith M, Barrera L, Chinen Y, et al. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12(6):931-45. [ Links ]
36. Kanazawa T, Yasunaga Y, Ikuta Y, Harada A, Kusaka O, Sukegawa K. Femoral head dysplasia in Morquio disease type A: bilateral varus osteotomy of the femur. Acta Orthop Scand. 2001;72(1):18-21. [ Links ]
37. Aranzadi E, Miramar MD, Cesar MA, Escanero JF. Evaluación del test del azul de dimetilmetileno (DMB) en el screening de las mucopolisacaridosis y su comparación con la prueba del cetilpiridinio (CPC). Rev Diagn Biol. 2006;55(1):17-21. [ Links ]
38. Romero R.E, Gutierrez OL, Castro SA, Bolivar AC. Guía de manejo errores innatos del metabolismo Bogotá : Hospital La Victoria; 2011. p. 12. [ Links ]
39. Chuang CK, Lin SP, Chung SF. Diagnostic screening for mucopolysaccharidoses by the dimethylmethylene blue method and two dimensional electrophoresis. Zhonghua Yi Xue Za <Zhi (Taipei). 2001;64(1):15-22. [ Links ]
40. Romero R.E, Gutierrez OL, Castro SA, Bolivar AC. Guía de manejo errores innatos del metabolismo Bogotá : Hospital La Victoria; 2011. p. 21-32. [ Links ]
41. Meikle PJ, Ranieri E, Simonsen H, Rozaklis T, Ramsay SL, Whitfield PD, et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics. 2004;114(4):909-16. [ Links ]
42. Khaliq T, Sadilek M, Scott CR, Turecek F, Gelb MH. Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: application to screening newborns for mucopolysaccharidosis IVA. Clin Chem. 2011;57(1):128-31. [ Links ]
43. Tomatsu S, Okamura K, Taketani T, Orii KO, Nishioka T, Gutierrez MA, et al. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr Res. 2004;55(4):592-7. [ Links ]
44. Chamoles NA, Blanco MB, Gaggioli D, Casentini C. Hurler-like phenotype: enzymatic diagnosis in dried blood spots on filter paper. Clin Chem. 2001;47(12):2098-102. [ Links ]
45. Guo YB, Ai Y, Zhao Y, Tang J, Jiang WY, Du ML, et al., Rapid prenatal genetic diagnosis of a fetus with a high risk for Morquio A syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2012;29(2):126-30. [ Links ]
46. National MPS Society. Guía Para Entender el Síndrome de Morquio, Durham: M. Society; 2011. p. 2-23. [ Links ]
47. Suárez Obando F, Zarante Montoya I. Aspectos clínicos y manejo integral del síndrome de Morquio. Univ Méd. 2007;48(2):166-74. [ Links ]
48. Tomatsu S, Montaño AM, Ohashi A, Gutierrez MA, Oikawa H, Oguma T, et al. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet. 2008;17(6):815-24. [ Links ]
49. Meza Cabrera IA, Álvarez J. Sindrome de Morquio. A propósito de un Caso. Revista Fac Cien Sal. 2010;12(1):41-3. [ Links ]
