










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.26 no.3 Bicaramanga Sept./Dec. 2013
Miastenia Gravis: una visión actual de la
enfermedad
Sergio Gómez*
Yelitza Álvarez**
Jorge Andrés Puerto***
* Estudiante de II nivel de Medicina. Oficial local de evaluación y desarrollo científico de la Fundación estudiantil de investigación médica (SEIMEDUIS). Escuela de Medicina. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
**Estudiante de IV nivel de Medicina. Oficial local de educación médica de la Fundación estudiantil de investigación médica (SEIMED-UIS). Escuela de Medicina. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
*** Estudiante de X nivel de Medicina. Director Científico de la Fundación estudiantil de investigación médica (SEIMED-UIS). Grupo de Inmunología y epidemiologia Molecular. Escuela de Medicina. Facultad de Salud. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
Correspondencia: Sr. Jorge Andrés Puerto F. Bloque 11-14 apto 301. Barrio Bucarica. Floridablanca. Colombia. Correo electrónico: jandres@hotmail.fr.
Artículo recibido el 19 de septiembre de 2013 y aceptado para publicación el 25 de noviembre de 2013
RESUMEN
Introducción: la miastenia gravis es una enfermedad autoinmune neuromuscular que afecta la placa motora. Su prevalencia es escasa, siendo mayor en países europeos y Estados Unidos. Pocas revisiones de tema se han publicado en Colombia y Latinoamérica sobre esta enfermedad. Objetivo: presentar una mirada actual y completa de la enfermedad a nivel mundial y de Colombia desde su epidemiologia, etiología, diagnóstico clínico, tratamiento y todo lo que el personal de salud y estudiantes deben saber. Metodología: se realizó una búsqueda en bibliotecas virtuales: Scopus, Google Académico y Medline con palabras claves: miastenia gravis, epidemiologia, Colombia, tratamiento, etiología y diagnóstico. Discusión: su principal característica es la debilidad muscular fluctuante, manifestada como fatiga en el músculo estriado esquelético. Existen varios abordajes para clasificarla, dependiendo de la edad de comienzo, el resultado de las pruebas serológicas y clínicamente por medio de las clasificaciones de Osserman y de la Myasthenia Gravis Foundation of America. Los métodos de diagnóstico se aplican dependiendo de las manifestaciones, sin embargo, aún no existe un método con especificidad del 100%. El tratamiento debe ser individualizado, basado en las características clínicas de cada paciente y dependiente del estadio de la enfermedad. Conclusión: a partir de la mejora en el conocimiento de la fisiopatología de la enfermedad, el diagnóstico y el tratamiento, se ha logrado reducir la mortalidad, así como, su subdiagnóstico, mejorando la calidad de vida de los pacientes y su posterior funcionalidad. (MÉD UIS. 2013;26(3):13-22).
Palabras Clave: Miastenia Gravis. Enfermedades Neuromusculares. Enfermedades Autoinmunes.
Myasthenia Gravis: a current vision of disease
ABSTRACT
Introduction: myasthenia gravis is a neuromuscular autoimmune disease which affects the motor plate. Its prevalence is low, being higher in European countries and the United States. Few reviews have been published in Colombia and Latin America about this disease. Aim: submit a current and complete look of the disease worldwide and Colombia from its epidemiology, etiology, clinical diagnosis, treatment and all health staff and students should know about this disease. Methods: we performed a search in virtual libraries: Scopus, Google Scholar and Medline with keywords: myasthenia gravis, epidemiology, Colombia, treatment, etiology, diagnosis. Discussion: The main feature is the fluctuating muscle weakness, manifested as fatigue in striated skeletal muscle. There are several approaches to classify, depending on the age of onset, serological test results and clinically by Osserman classifications and the Myasthenia Gravis Foundation of America MGFA. Diagnostic methods are applied depending on the clinical manifestations, however, there is still no method with specificity of 100%. Treatment must be individualized based on the clinical characteristics of each patient and dependent on the stage of the disease Conclusion: Through improved knowledge of the disease, diagnosis and treatment, mortality has been reduced, as well as underdiagnosis, improving the quality of life of patients and their subsequent function. (MÉD UIS. 2013;26(3):13-22).
Keywords: Myasthenia Gravis. Neuromuscular Diseases. Autoimmune Diseases.
INTRODUCCIÓN
La contracción normal de las fibras musculares estriadas esqueléticas se realiza por medio de nervios motores, estos se ramifican en el interior del tejido conectivo del perimisio, originando numerosas terminaciones sinápticas denominadas placa motora. Cuando el potencial de acción llega a la unión neuromuscular se libera acetilcolina de los botones terminales; esta es difundida hacia el espacio sináptico y se une a receptores nicotínicos postsinápticos (canales iónicos), lo que permite despolarizar la membrana y originar un potencial de placa motora. Si este alcanza una despolarización umbral, se dispersa un potencial de acción a lo largo de toda la fibra muscular, originando la contracción del músculo. La acetilcolina es hidrolizada del espacio sináptico por la acetilcolinesterasa1.
La Miastenia Gravis (MG) es un trastorno autoinmune caracterizado por la presencia de anticuerpos contra los receptores de acetilcolina o de proteínas involucradas en la región postsináptica de la placa motora, debido a esto los potenciales de placa son insuficientes para generar potenciales de acción en las fibras musculares, resultando en una falla en la trasmisión neuromuscular1-3. La enfermedad se caracteriza por presentar fatigabilidad y debilidad fluctuante en el músculo esquelético que tiende a mejorar en estados de reposo; por lo general afecta a grupos musculares determinados, siendo en sus inicios más afectados los oculares y en su transcurso se van adicionando los músculos bulbares, axiales, de extremidades e incluso los respiratorios en situaciones más graves como la crisis miasténica2,4-6. No es una neuropatía común, pero una vez manifestada en el paciente afecta notoriamente su calidad de vida, por lo que es necesario el conocimiento de las particularidades que la caracterizan.
En la literatura mundial se encuentran múltiples textos que abordan uno o varios aspectos sobre esta enfermedad. Sin embargo, se hace necesaria una revisión que involucre todos los aspectos de la misma, incluyéndose parámetros de epidemiología, etiología, manifestaciones clínicas, diagnóstico y tratamiento de manera concreta. Por lo tanto, el objetivo de la presente revisión es abordar y actualizar de manera completa y sintetizada los aspectos de mayor importancia de la enfermedad desde distintos enfoques útiles para la comunidad médica. Además se pretende destacar la importancia de su correcto diagnóstico en el incremento y detección de los casos, evitando confundirla con otras neuropatías musculares.
METODOLOGÍA
Se realizó una búsqueda en bases de datos: Scopus, Medline y Google Académico, desde el mes de febrero del año 2013 hasta el mes de septiembre del mismo año, con palabras claves incluyera: miastenia gravis, epidemiología, Latinoamérica, Colombia, etiología, manifestaciones clínicas, diagnostico. Se encontraron 150 artículos de revistas en línea, de los cuales se seleccionaron 81 para la revisión. Criterios de inclusión: artículos publicados a partir del año 2000, excepto los que contenían definiciones de la enfermedad que datan desde el año 1934, artículos originales y de revisión de tema que a juicio de los autores fueran pertinentes para la revisión. Criterios de exclusión: se tuvieron en cuenta artículos que desde su enfoque no aportaran información esencial y útil para hacer la revisión y se alejaran del objetivo planteado por los autores.
EPIDEMIOLOGÍA
La MG no es un trastorno frecuente, sin embargo desde la década de los 80 se describen en la literatura incrementos anuales en su incidencia en promedio de 0,53 por 100 000 personas. Su prevalencia es muy variable y fluctúa de 1,5 hasta 17,9 por 100 000 habitantes7. La enfermedad tiene una mayor prevalencia en Estados Unidos y países Europeos, siendo Pavia, una provincia italiana, el lugar con más casos reportados hasta el momento: 24 por cada 100 000 habitantes8. La baja prevalencia en algunos países latinoamericanos con clima tropical es similar entre estos mismos9,10.
América y Europa registran pocos casos en niños menores de 15 años, sin embargo en, estudios realizados en países asiáticos se ha encontrado una mayor frecuencia de casos en la población juvenil. En China el 39 a 50% de los casos fueron en niños11; en Japón, el 7% de los pacientes tenían de cero a cuatro años de vida y el 80,6% de estos presentaba Miastenia Gravis ocular (MGo), un pico considerable en la distribución de la enfermedad respecto a la edad12.
La población que presenta un comienzo tardío de la enfermedad o después de los 50 años, es la más afectada; su incidencia ha aumentado en los últimos años significativamente en comparación de otros estadios de vida12-4. El porcentaje de casos presenta un declive después de los 80 años15,16.
En Colombia, solo se ha publicado un estudio realizado en el departamento de Antioquia, "Prevalence of Myasthenia Gravis in Antioquia, Colombia", quel mostró una proporción de mujeres a hombres de 3,7:19, lo que concuerda con lo reportado en la literatura mundial respecto a la mayor presencia de la enfermedad en el sexo femenino.
En mujeres tiende a manifestarse de los 20 a 40 años mientras que en hombres se presenta generalmente después de los 50 años8,17. Sin embargo algunos estudios muestran un segundo pico en el sexo femenino a partir de los 60 años13.
ETIOLOGÍA
Se han descrito múltiples hipótesis que tratan de explicar el mecanismo por el que se desarrolla la MG, sin embargo, dentro de las más estudiadas en la actualidad, se encuentran las relacionadas con la respuesta inmune y los cambios genéticos que ocurren en las células que participan en ella18.
La hipótesis autoinmune se fundamenta a partir de la producción de anticuerpos (Ac) contra receptores de Acetilcolina (AchR) en ciertos tipos de pacientes19. Esta variante de la enfermedad, se acompaña de cambios funcionales y estructurales en el timo, como timoma en el 15% de los pacientes o la presencia de hiperplasia timica en el 60% de la población afectada con este tipo de patología20. En el timo se desarrolla el ambiente adecuado para una respuesta autorreactiva de células B, células T y células presentadoras de antígenos contra AChR21. Adicional a esto, hay un aumento en el número de células mononucleares, así como IFN y e IL 4, demostrando la presencia de una respuesta Th1 y Th2. Las células Th1 aumentan el número de receptores CXC3, que son quimiorreceptores de la inflamación. Los receptores Th2 producen IL-4, IL-5, IL-9, IL-10 e IL-13, todo esto prolonga el estado inflamatorio del paciente22.
Una segunda hipótesis está relacionada con una elevada activación de marcadores de inflamación observados en pacientes con MG, lo que demuestra un sustento más, sobre la hipótesis de una respuesta crónica y sistémica que incluye diferentes subtipos de células T CD4+ 23. De las más estudiadas, se encuentran las células T reguladoras (Treg) CD4+ CD25+, ya que estas células tienen un papel importante en la homeostasis inmune y la autotolerancia24. Sin embargo, en un estudio realizado en pacientes con MG comparados con pacientes sanos, no hubo una significancia estadística en el número de células Treg CD4+ CD25+25. No obstante, se encontró que el factor de transcripción Forkhead box protein P3 (FOXP3), el cual se expresa en células Treg CD4+ CD25+ y cuya función es ser el más importante regulador en la conversión de células T CD4+ CD25- en poblaciones de linfocitos reguladores funcionales Treg CD4+, estaba significativamente disminuido26.
Una tercera hipótesis se relaciona con una producción de Ac contra la proteína quinasa transmembrana postsináptica especifica del músculo, conocida en sus siglas en inglés como MUSK entre el 5 a 10% de los pacientes con MG27. Se ha demostrado que la IgG subclase 4 se relaciona con la severidad de la enfermedad sugiriendo actividad miastogénica. Esta subclase de Inmunoglobulina (Ig) se consideraba "benigna" hasta ahora. Sin embargo, por medio de la purificación de IgG 4, de pacientes con MUSK, que se ligaron a uniones neuromusculares de ratones inmunodeficientes in vitro, les provocó parálisis. Análisis electrofisiológicos posteriores mostraron una reducción de la sensibilidad postsináptica de Acetilcolina (ACh), una depresión en la liberación de ACh a nivel presináptico durante altas tasas de actividad, todas juntas, causando debilidad o fatiga muscular28.
Estudios recientes han demostrado la presencia de interacción de factores genéticos y ambientales en la MG, dándole un carácter de multifactorialidad al igual que en otras enfermedades autoinmunes29,30. Además, se han encontrado polimorfismos sobre genes que se encuentran en la región cromosómica asociados a susceptibilidad de desarrollo de MG. Sin embargo, se asocian con otro tipo de enfermedades autoinmunitarias31.
MANIFESTACIONES CLÍNICAS
La característica principal de la MG es la debilidad muscular fluctuante. Se manifiesta principalmente como fatiga, que empeora con actividades repetitivas y mejora con el reposo. Esta se ve agravada por la exposición al calor, las infecciones y el estrés32. La debilidad del músculo esquelético implica grupos específicos, siendo su distribución generalmente bulbar, ocular, en extremidades proximales, cuello, y en algunos pacientes en crisis, incluye los músculos respiratorios33.
La debilidad muscular ocular, clasificada en el grupo Adulto 1 de Osserman y Clase I según la Myasthenia Gravis Foundation of America (MGFA). (Ver Figura 1), es el síntoma inicial más común de la MG, ocurriendo aproximadamente en el 85% de los pacientes32. Es provocada por la debilidad de más de un musculo extraocular, siendo el más comprometido el recto medial. Se presenta como ptosis fluctuante y diplopía33. Solo un pequeño porcentaje de pacientes presenta solo ptosis o diplopía34. La ptosis puede ser unilateral o bilateral, la unilateral es generalmente inducida por la mirada sostenida hacia arriba durante 30 segundos o más. Si es bilateral, la ptosis puede ser lo suficientemente grave como para ocluir totalmente la visión. La debilidad no sigue ningún patrón de nervio específico o afectación de los músculos, lo que la distingue de otros trastornos como la parálisis oculomotora, parálisis vertical de la mirada u oftalmoplejía internuclear35. Posteriormente afecta la musculatura facial y bulbar, produciendo parálisis y disfagia, que genera dificultad para masticar debido a la debilidad en el músculo masetero, esta última puede llegar al extremo de incapacitar el músculo, evitando que este pueda mantener la mandíbula cerrada4,35,36.
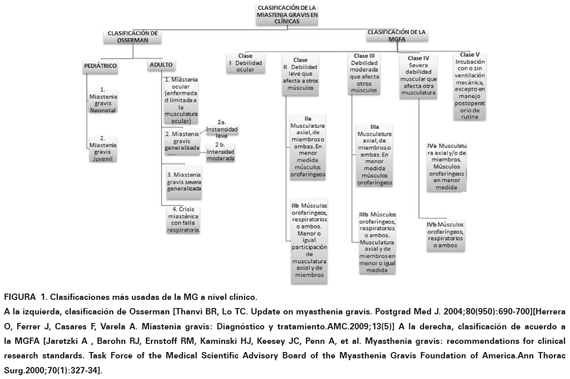
En el desarrollo posterior de la enfermedad el compromiso de las extremidades se manifiesta como debilidad muscular proximal, similar a otros trastornos miopáticos. Los miembros superiores tienden a resultar más afectados que los miembros inferiores. Sin embargo, la debilidad muscular distal puede ser otra manifestación37. Además, la capacidad reducida de los músculos extensores del cuello puede producir un "síndrome de cabeza caída"38. Como estadio final, se desarrolla Miastenia Gravis generalizada (MGg) presentándose en el 85% de los pacientes39. Afecta las extremidades, los músculos del cuello y el diafragma; cuando este último se afecta a tal punto que se necesita asistencia respiratoria mecánica, se dice que el paciente se encuentra en una crisis miasténica clasificada en el grupo Adulto 4 de Osserman y en Clase V según la MGFA (Ver Figura 1) siendo este el último estadio de la enfermedad40.
CRISIS MIASTÉNICA
El estadio más grave de la MG se denomina crisis miasténica, la cual es una causa reversible de la parálisis neuromuscular, que requiere de un diagnóstico precoz y asistencia respiratoria, pues los músculos comprometidos son de vital importancia4. La crisis miasténica es la condición final de la enfermedad. Se dice que hay crisis cuando existe insuficiencia respiratoria o extubación posoperatoria retrasada por timectomía por más de 24 horas. Esta condición lleva a una reducción del volumen corriente41. Sin embargo, al aparecer un aumento rápido y marcado en la debilidad bulbar y muscular distal sin existir insuficiencia respiratoria, debe ser también definido como crisis en un paciente miasténico42.
Entre los factores que desatan la crisis se cuentan infecciones respiratorias, sepsis, procedimientos quirúrgicos, aspiración, disminución rápida de la modulación inmune por medio de un tratamiento con corticoides, la exposición a los fármacos que pueden aumentar la debilidad miasténica y el estado de embarazo, este último debido a la brusca caída en los niveles séricos de la alfa-fetoproteína43. La crisis no es fatal si se suministra a tiempo el soporte respiratorio y se realiza una inmunoterapia para reducir la debilidad miasténica de los músculos de las vías respiratorias41.
El manejo respiratorio de estos pacientes es muy difícil debido a la naturaleza fluctuante de la enfermedad42. Sin embargo, mediante la mejora en la evaluación de los cuidados críticos, la tasa de mortalidad de la crisis miasténica ha disminuido, ubicándose entre 4 y 8% en los últimos años44.
CLASIFICACIÓN DE LA MIASTENIA GRAVIS
De acuerdo a pruebas serológicas
Uno de los protocolos más usados para confirmar el diagnóstico de MG es la medición de Ac contra AChR por medio de radioinmuno ensayo45. Si la prueba serológica es positiva para éstos, la enfermedad se clasifica en MG seropositiva, la cual es la forma de presentación más frecuente y aborda un 85% de los casos46-48. En un 15 a 20% de los pacientes no se detectan Ac contra AChR y la enfermedad es clasificada en MG seronegativa. En la mayor parte de este porcentaje son encontrados altos niveles de Ac-MUSK. El MUSK es un polipéptido transmembrana ubicado en la placa motora que mantiene su integridad agrupando a los AChR en la membrana muscular. En aproximadamente un 8% no es encontrado ningún tipo de Ac en el suero para llevar a cabo el diagnóstico, esto es conocido como MG-doble seronegativa48.
CLASIFICACIÓN CLÍNICA
Se clasifica teniendo en cuenta los síntomas, la evolución y el estadio de la enfermedad. Entre las más referenciadas se encuentra modificaciones de la hecha por Osserman2,17 y la creada por la MGFA49.
La MGFA cataloga a la enfermedad en cinco clases: la primera se delimita solo a musculatura ocular; en la segunda, tercera y cuarta se incluye tanto debilidad ocular como de otros grupos musculares, cada una de ellas con una subclasificación A y B. En cada subclase hay un tipo de musculatura más afectado, sin embargo el otro puede involucrarse en menor o igual medida, de esta manera en la subclase A hay afección predominante de la musculatura axial y/o de extremidades, y en la subclase B es predominante en músculos orofaríngeos, respiratorios o incluso ambos. La quinta clase hace referencia a crisis miasténica5.
De acuerdo a la edad de comienzo
Cuando la MG comienza antes de los 50 años es de inicio temprano, por lo general se presenta más en mujeres. Los pacientes manifiestan Ac contra AChR y la severidad de la enfermedad tiende a estar en sus primeras fases. Después de los 50 años es de inicio tardío, más frecuente en hombres y tiende a estar más avanzada encontrándose incluso crisis miasténicas5, 47, 50.
MÉTODOS DE DIAGNÓSTICO CLÍNICO
El diagnóstico de la MG se lleva a cabo mediante una revisión de los síntomas y manifestaciones que presenta el paciente y de su historia clínica. Para confirmar, se realizan algunas pruebas a nivel clínico, otras que permitan detectar la presencia de Ac y tests electrofisiológicos. El mayor reto que se presenta es diferenciar la condición de otros trastornos autoinmunes de la transmisión neuromuscular como el síndrome miasténico de Lambert-Eaton, síndrome de Guillain Barré, esclerosis lateral amiotrófica, entre otras50.
Prueba de edrofonio
El cloruro de edrofonio (Tensilón®) es un fármaco anticolinesterásico muy usado a nivel clínico para diagnosticar la MG. Su mecanismo de acción es inmediato, por medio de inhibición competitiva bloquea a la colinesterasa, una enzima encargada de descomponer la ACh en la hendidura sináptica, permitiendo que el neurotransmisor llegue más fácilmente a un receptor muscarínico; su efecto persiste por cinco a diez minutos aproximadamente1,6. La prueba tiene una sensibilidad del 86% para MGo y del 95% para MGg, aunque su sensibilidad es alta se han reportado falsos negativos en aproximadamente un 20% de los casos2. Es más efectiva en pacientes con debilidad en los músculos extraoculares, es decir con MGo. Se administra vía intravenosa y es considerada como positiva cuando se observa una mejora notoria a medida que se aumenta la dosis requerida sin necesidad de que llegue a una dosificación máxima. Debido a su potencia muscarínica se pueden presentar efectos secundarios tales como sudoración, salivación, náuseas, diarrea, fasciculación muscular, entre otros1,2,5,6.
Prueba de la bolsa de hielo
Es usada como una alternativa para los pacientes con comorbilidades cardíacas y respiratorias a los cuales no se les puede administrar cloruro de edrofonio. Es útil solo en pacientes que manifiestan ptosis palpebral. La prueba consiste en aplicar bolsas de hielo sobre los ojos cerrados del paciente por aproximadamente dos a cinco minutos1,5, la ptosis debe mejorar y permanecer así por un tiempo corto. Con una sensibilidad del 80% su especificidad es aún desconocida, sin embargo se ha reportado que al aplicar la prueba en pacientes que presentan ptosis sin tener MG éstos no manifiestan mejoría51.
Prueba de medición serológica de anticuerpos
La búsqueda de Ac contra AChR es la prueba más específica para MG, reportándose falsos positivos solo en 5%5, con una sensibilidad del 85% para MGo y de un 50% para MGg6,17,52. Puede resultar seronegativa para AChR, pero no se rechaza el diagnóstico si son encontrados otros como los Ac contra MUSK presentes en el 40% de los casos seronegativos con MGg5, o los antimúsculo estriados, detectados en un 80% en pacientes con anomalías tímicas6.
PRUEBAS ELECTROFISIOLÓGICAS
Estimulación repetitiva del nervio
En la estimulación repetitiva del nervio se evalúa la transmisión neuromuscular. Se hace una estimulación supramáxima en un nervio motor a una baja frecuencia, en promedio de 3 Hz45,52. La respuesta normal del potencial de acción compuesto manifestado en el músculo es una disminución en un 8% desde la primera a quinta estimulación, un decremento igual o mayor a un 10% es un diagnóstico positivo para la enfermedad52,53. La prueba tiene una sensibilidad del 90% para MGg y del 60% para MGo53.
Electromiografía de fibra única
Se emplea un electrodo de 25 μm de diámetro capaz de identificar el potencial de acción en una fibra muscular, para llevar a cabo la prueba se estudian dos fibras musculares activadas por una unidad motora52. El criterio usado para evaluar al paciente es el "Jitter"54 término inglés para fluctuación. Este mide el tiempo que tarda el potencial de placa para llegar al umbral, en variación de intervalos de tiempo entre la primera fibra y la segunda52. El jitter es una medición del factor de seguridad de la función neuromuscular, cuando está incrementado se considera la presencia de MG45,52.
Generalmente el músculo elegido tiende a ser el extensor común de los dedos en MGg y el occipitofrontal u orbicular de los párpados en MGo52- 54. Sin embargo, dependiendo de las manifestaciones clínicas se pueden elegir otros más convenientes. Se ha demostrado que el masetero puede contribuir efectivamente al diagnóstico de la enfermedad, sobre todo cuando existe compromiso bulbar55. Es la prueba de mayor sensibilidad, de 95 a 99%, sobre todo si es MGg53, pero su especificidad es muy baja debido a que puede detectar cualquier anomalía neuromuscular52.
TRATAMIENTO
El tratamiento de la MG debe ser individualizado para cada paciente y basarse en las características clínicas del paciente, entre las cuales incluye la distribución, duración y gravedad de la debilidad, además en los riesgos y complicaciones del tratamiento relacionados con la edad, sexo, comorbilidades médicas y el resultante deterioro funcional, igualmente se debe tener en cuenta la presencia de un timoma32.
TRATAMIENTO SINTOMÁTICO POR MEDIO DE
INHIBIDORES DE LA ACETILCOLINESTERASA
El manejo farmacológico de los síntomas de la MG incluye los inhibidores de la acetilcolinesterasa, especialmente la piridostigmina, la cual bloquea la acetilcolinesterasa presente en la matriz extracelular de la membrana de la placa terminal plegada, mejorando la exposición de la ACh sobre los receptores afectados56. Por consiguiente los inhibidores de la acetilcolinesterasa de acción prolongada por sí solos pueden proporcionar un beneficio funcional satisfactorio sin los efectos secundarios potenciales de los moduladores inmunes en pacientes con MGo y MGg32.
Su uso en los primeros estadios de MG ha demostrado ser efectivo, sin embargo después de meses de iniciado el tratamiento se van requiriendo dosis cada vez mayores para mantener el resultado, finalmente el efecto disminuye, incluso al utilizarse a dosis máximas; de ser así, este tratamiento debe ser descontinuado36. La dosis óptima y la duración del tratamiento se precisan por el equilibrio entre la mejoría clínica del paciente y los efectos adversos. Esto varía con el tiempo y depende de otros tipos de tratamiento que se estén llevando a cabo para inhibir la respuesta autoinmune subyacente57.
Debe haber una medición adecuada de cada dosis pues el exceso de dosificación puede aumentar potencialmente la debilidad atribuible al bloqueo de despolarización en las uniones neuromusculares afectadas32. Existen varios estudios que sugieren que el uso de estos medicamentos debe ser detenido durante 48 hasta 72 horas, con el fin de evitar que la membrana postsináptica se sobrecargue, lo que permite la expresión de nuevos receptores de ACh en la placa motora, para más tarde reincorporar los medicamentos por sonda nasogástrica38.
TRATAMIENTO INMUNOMODULADOR A CORTO PLA ZO
USANDO PLASMAFÉRESIS E INMUNOGLOBULINA
INTRAVENOSA
El intercambio plasmático terapéutico o plasmaféresis es un procedimiento diseñado para extraer el plasma de la sangre sin comprometer células sanguíneas. El procedimiento se utiliza para eliminar el exceso de Ig y citoquinas en la sangre en diversas situaciones clínicas58. Este reduce el número de Ac y alivia los síntomas de forma temporal ya que no se puede evitar la resíntesis de estos Ac. Además, existen varios efectos adversos de este procedimiento incluyendo hipotensión, coagulopatía y las complicaciones relacionadas con el catéter lo que ha puesto en duda en cuanto a si se debe utilizar de forma rutinaria o sólo en casos seleccionados59.
La Ig intravenosa es un procedimiento eficaz para el tratamiento de enfermedades autoinmunes como la MG. Utiliza Ac purificados a partir de sangre humana. Se trata de aislar Ig a partir de plasma humano agrupado por crioprecipitación con etanol, para ser luego administrado al paciente60. Esta técnica es considerada segura, aun así, se han encontrado eventos adversos como fiebre, náuseas y dolor de cabeza debido a la Ig intravenosa, aunque estos eventos son autolimitados y menos graves que con la plasmaféresis61. Además, pueden existir complicaciones como coagulopatías debido al incremento en la viscosidad de la sangre y otras relacionadas con los grandes volúmenes de la preparación administrada62.
TRATAMIENTO INMUNOMODULADOR CRÓNICO A
LARGO PLA ZO CON GLUCOCORTICOIDES Y OTROS
FÁRMACOS INMUNOSUPRESORES
Los corticoides fueron los primeros medicamentos inmunosupresores utilizados en MG. Uno de estos, la prednisona se usa cuando los síntomas no son controlados apropiadamente por inhibidores de colinesterasa63. Una buena respuesta puede conseguirse con dosis iniciales altas, para luego disminuir a dosis más bajas con el fin de mantener el resultado, debido a que altas dosis de prednisona pueden provocar exacerbación de la debilidad, durando esta varios días64. En caso de sospecha de una exacerbación grave de la enfermedad, se recomienda realizar tratamiento con plasmaféresis o Ig intravenosa para prevenir y reducir la gravedad de la debilidad provocada por los corticoides e incitar una respuesta más rápida. La prednisona oral podría ser más eficaz que los fármacos anticolinesterásicos en pacientes con MGo65,66.
La MG también puede ser tratada con agentes inmunosupresivos no esteroideos, por ejemplo, el micofenolato mofetilo, que bloquea selectivamente la síntesis de purinas, suprimiendo de ese modo la proliferación tanto de las células T como de células B. También se hace uso del Tacrolimus, un agente inmunosupresor que ha sido utilizado con éxito para tratar la MG usando bajas dosis. Este tratamiento induce poca nefrotoxicidad, pero al igual que otros agentes inmunosupresores, también tiene el potencial de efectos secundarios graves1.
Otro medicamento utilizado, la ciclofosfamida, es una ciclosporina que bloquea la síntesis de los receptores de IL-2 y otras proteínas fundamentales para la función de células T CD4+ 67. Se administra vía intravenosa u oral y es un tratamiento práctico y efectivo, pues un alto porcentaje de los pacientes se tornan asintomáticos durante el primer año de tratamiento. Existen efectos secundarios indeseables como la pérdida de cabello, náuseas, vómito, anorexia y decoloración de la piel, por tanto, se restringe su uso para el manejo de los pacientes que no manifiestan mejoría con otros tratamientos inmunosupresores1.
Los pacientes resistentes a otras terapias han sido tratados con éxito con este medicamento en combinación con trasplante de médula ósea o con rituximab, este último es un anticuerpo monoclonal contra el marcador de superficie de células B CD20+ 68.
Todo tratamiento inmunomodulador tiene el potencial de efectos secundarios, siendo unos más graves que otros, por tanto debe tenerse en cuenta la etapa de la enfermedad y las características clínicas del paciente antes de iniciar el tratamiento, siendo los inhibidores de la acetilcolinesterasa los más adecuados a administrar en estadios de MGo y MGg leve y la ciclofosfamida como opción final ante una mejoría nula con otros tratamientos inmunosupresores.
TRATAMIENTO QUIRÚRGICO
La timectomía es el tratamiento predilecto para los pacientes con MG que tienen un timoma69. A pesar del avance en los tratamientos, la timectomía sigue siendo una parte integral a la hora de abordar la MG70. A largo plazo, este procedimiento es superior al tratamiento conservador con respecto a la supervivencia global, mejoría clínica y tasa de remisión71. Aun así, la eficacia a largo plazo y el momento óptimo para esta intervención en miastenia juvenil siguen siendo controvertidos, por lo que no es recomendado como primera opción en este tipo de miastenia72.
La eficacia de la cirugía depende de la escisión completa del timo. Existen varias técnicas quirúrgicas para el tratamiento de la MG, como la timectomía transcervical, la timectomía transesternal y la cirugía toracoscópica asistida por video73, se ha demostrado que esta última tiene una alta eficacia con menos efectos adversos y mejores resultados estéticos74. Por otra parte, se comprobó que la timectomía toracoscópica es una estrategia costo-efectiva en comparación con el tratamiento farmacológico de MG sin timoma75, además, de ser eficaz en el alivio de los síntomas de la pseudo-obstrucción colónica en pacientes miasténicos con timomas recurrentes76.
La radioterapia tiene un papel reconocido como un tratamiento adyuvante a la cirugía en caso de un timoma y se ha mantenido como el único tratamiento disponible, con o sin quimioterapia, en los tumores considerados irresecables. La resecabilidad y la respuesta a la quimioterapia en pacientes inoperables parecen ser los factores que predicen el resultado del timoma77.
Se sabe que la recurrencia de un timoma invasivo es un acontecimiento frecuente, puede ocurrir entre el 10 al 30% de los pacientes en un intervalo muy amplio posterior a la timectomía78, por ejemplo un caso se reportó luego de 32 años79. En timomas recurrentes la cirugía reiterativa, una nueva técnica de resección quirúrgica, se ha demostrado como método eficaz. Hasta la fecha, es posible decir que la tasa de curación de esta es superior a la de la radioterapia y la quimioterapia80,81.
Finalmente, es recomendable iniciar un tratamiento de rehabilitación con el fin de aliviar los síntomas y mejorar la función en esta enfermedad para garantizar el regreso a las actividades de la vida diaria del paciente. Para determinar la intensidad y duración del ejercicio debe tomarse en cuenta la etapa de la enfermedad en la que se encuentre y el estado de salud del individuo. Debe abordarse con un enfoque interdisciplinario, que cubra todos los aspectos de importancia en el tratamiento, incluyendo terapia respiratoria y rehabilitación física. Esta última será un factor positivo para la recuperación de la fuerza muscular con el fin de hacer al individuo lo más funcional posible. Posterior a esta, debe acostumbrarse al individuo a las nuevas formas de realizar sus actividades diarias siendo necesaria una terapia ocupacional, que mida los límites de estas. En circunstancias especiales puede ser requerida terapia del habla, en el caso de una traqueotomía, o una serie de consultas con un psicólogo60.
CONCLUSIONES
Los avances en el conocimiento de la epidemiología, fisiopatología, la mejora en distintos métodos diagnósticos junto con la amplia variedad de tratamientos disponibles y el aumento en disponibilidad de recursos médicos para detectar la MG han permitido reducir su mortalidad, mejorando la calidad de vida de los pacientes y su posterior funcionalidad. Sin embargo, la falta de actualización y profundización del personal de salud sobre la MG, ha conllevado a confundirla con otras enfermedades, aumentando la probabilidad de subdiagnóstico clínico, generando graves consecuencias en el paciente afectado con esta patología. Sumado a esto se encuentra la falta de estudios en Colombia acerca de la MG, lo que pudiera impedir esclarecer las posibles diferencias o semejanzas en la presentación global de esta enfermedad respecto a la población colombiana.
Pudiera todo lo anteriormente expuesto superarse con capacitaciones periódicas al personal de salud no solamente sobre MG, sino también, las enfermedades neuromusculares que pueden ser confundidas con esta y que debido a su rápida progresión comprometen la vida del paciente en días e incluso horas.
AGRADECIMENTOS
Agradecemos a la Fundación estudiantil de investigación médica de la Universidad Industrial de Santander SEIMED-UIS, por la realización de este trabajo en el marco de la formación científica estudiantil en el pregrado del año 2013.
REFERENCIAS BIBLIOGRAFICAS
1. Scherer K, Bedlack RS, Simel DL. Does this patient have myasthenia gravis?. JAMA. 2005;293(15):1906-14. [ Links ]
2. Herrera O, Ferrer J, Casares F, Varela A. Miastenia gravis: diagnóstico y tratamiento. AMC. 2009;13(5). [ Links ]
3. Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116(11):2843-54. [ Links ]
4. Chaudhuri A, Behan PO. Myasthenic crisis. QJM. 2009;102(2):97-107. [ Links ]
5. Jayam Trouth A, Dabi A, Solieman N, Kurukumbi M, Kalyanam J. Myasthenia gravis: a review. Autoimmune Dis. 2012;2012:1-10. [ Links ]
6. Juel VC, Massey JM. Myasthenia gravis. Orphanet J Rare Dis. 2007;2:44. [ Links ]
7. Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010;10:46. [ Links ]
8. Montomoli C, Citterio A, Piccolo G, Cioccale R, Ferretti VV, Fratti C, et al. Epidemiology and geographical variation of myasthenia gravis in the province of Pavia, Italy. Neuroepidemiology. 2012;38(2):100-5. [ Links ]
9. Sánchez JL, Uribe CS, Franco AF, Jiménez ME, Arcos-Burgos OM, Palacio LG. Prevalence of myasthenia gravis in Antioquia, Colombia. Rev Neurol. 2002;34(11):1010-2. [ Links ]
10. Aguilar Ade A, Carvalho AF, Costa CM, Fernandes Jm, D'Almeida JA, Furtado LE, et al. Myasthenia gravis in Ceará, Brazil: clinical and epidemiological aspects. Arq Neuropsiquiatr. 2010;68(6):843-8. [ Links ]
11. Wong V, Hawkins BR, Yu YL. Myasthenia gravis in Hong Kong Chinese. 2. Paediatric disease. Acta Neurol Scand. 1992;86(1):68-72. [ Links ]
12. Murai H, Yamashita N, Watanabe M, Nomura Y, Motomura M, Yoshikawa H, et al. Characteristics of myasthenia gravis according to onset-age: Japanese nationwide survey. J Neurol Sci. 2011;305(1-2):97-102. [ Links ]
13. Cetin H, Fülöp G, Zach H, Auff E, Zimprich F. Epidemiology of myasthenia gravis in Austria: rising prevalence in an ageing society. Wien Klin Wochenschr. 2012;124(21-22):763-8. [ Links ]
14. Lai CH, Tseng HF. Nationwide population-based epidemiological study of myasthenia gravis in taiwan. Neuroepidemiology. 2010;35(1):66-71. [ Links ]
15. Oöpik M, Kaasik AE, Jakobsen J. A population based epidemiological study on myasthenia gravis in Estonia. J Neurol Neurosurg Psychiatry. 2003;74(12):1638-43. [ Links ]
16. Vincent A, Clover L, Buckley C, Grimley Evans J, Rothwell PM; UK Myasthenia Gravis Survey. Evidence of underdiagnosis of myasthenia gravis in older people. J Neurol Neurosurg Psychiatry. 2003;74(8):1105-8. [ Links ]
17. Thanvi BR, Lo TC. Update on myasthenia gravis. Postgrad Med J. 2004;80(950):690-700. [ Links ]
18. Thiruppathi M, Rowin J, Li Jiang Q, Sheng JR, Prabhakar BS, Meriggioli MN. Functional defect in regulatory T cells in myasthenia gravis. Ann N Y Acad Sci. 2012;1274:68-76. [ Links ]
19. Guyon T, Levasseur P, Truffault F, Cottin C, Gaud C, Berrih-Aknin S. Regulation of acetylcholine receptor alpha subunit variants in human myasthenia gravis. Quantification of steady-state levels of messenger RNA in muscle biopsy using the polymerase chain reaction. J Clin Invest. 1994;94(1):16-24. [ Links ]
20. Berrih S, Morel E, Gaud C, Raimond F, Le Brigand H, Bach JF. Anti-AChR antibodies, thymic histology, and T cell subsets in myasthenia gravis. Neurology. 1984;34(1):66-71. [ Links ]
21. Leprince C, Cohen-Kaminsky S, Berrih-Aknin S, Vernet-Der Garabedian B, Treton D, Galanaud P, et al. Thymic B cells from myasthenia gravis patients are activated B cells. Phenotypic and functional analysis. J Immunol. 1990;145(7):2115-22. [ Links ]
22. Akdis M, Burgler S, Crameri R, Eiwegger T, Fujita H, Gomez E, et al. Interleukins, from 1 to 37, and interferon- y: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2011;127(3):701-21. e1-70. [ Links ]
23. Gradolatto A, Nazzal D, Foti M, Bismuth J, Truffault F, Le Panse R, et al. Defects of immunoregulatory mechanisms in myasthenia gravis: role of IL-17. Ann N Y Acad Sci. 2012;1274:40-7. [ Links ]
24. Hori S, Takahashi T, Sakaguchi S. Control of autoimmunity by naturally arising regulatory CD4+ T cells. Adv Immunol. 2003;81:331-71. [ Links ]
25. Battaglia A, Di Schino C, Fattorossi A, Scambia G, Evoli A. Circulating CD4+CD25+ T regulatory and natural killer T cells in patients with myasthenia gravis: a flow cytometry study. J Biol Regul Homeost Agents. 2005;19(1-2):54-62. [ Links ]
26. Zhang Y, Wang HB, Chi LJ, Wang WZ. The role of FoxP3+CD4+CD25hi Tregs in the pathogenesis of myasthenia gravis. Immunol Lett. 2009;122(1):52-7. [ Links ]
27. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7(3):365-8. [ Links ]
28. Klooster R, Plomp JJ, Huijbers MG, Niks EH, Straasheijm KR, Detmers FJ, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain. 2012;135(Pt 4):1081-101. [ Links ]
29. Vandiedonck C, M Giraud, Garchon HJ. Genetics of autoimmune myasthenia gravis: the multifaceted contribution of the HLA complex. J Autoimmun. 2005;25 Suppl:6-11. [ Links ]
30. Cavalcante P, Cufi P, Mantegazza R, Berrih-Aknin S, Bernasconi P, Le Panse, R. Etiology of myasthenia gravis: innate immunity signature in pathological thymus. Autoimmun Rev. 2013;12(9):863-74. [ Links ]
31. Skeie GO, Pandey JP, Aarli JA, Gilhus NE. TNFA and TNFB polymorphisms in myasthenia gravis. Arch Neurol. 1999;56(4):457-61. [ Links ]
32. Grob D, Arsura EL, Brunner NG, Namba T. The course of myasthenia gravis and therapies affecting outcome. Ann N Y Acad Sci. 1987;505:472-99. [ Links ]
33. Juel VC, Massey JM. Autoimmune Myasthenia Gravis: Recommendations for Treatment and Immunologic Modulation. Curr Treat Options Neurol. 2005; 7(1):3-14. [ Links ]
34. Roh HS, Lee SY, Yoon JS. Comparison of clinical manifestations between patients with ocular myasthenia gravis and generalized myasthenia gravis. Korean J Ophthalmol. 2011;25(1):1-7. [ Links ]
35. DiMauro S, Garone C. Myasthenia gravis. 2013. [ Links ]
36. Thomas CE, Mayer SA, Gungor Y, Swarup R, Webster EA, Chang I, et al. Myasthenic crisis: clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology. 1997;48:1253-60. [ Links ]
37. Werner P, Kiechl S, Löscher W, Poewe W, Willeit J. Distal myasthenia gravis frequency and clinical course in a large prospective series. Acta Neurol Scand. 2003;108(3):209-11. [ Links ]
38. Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle and Nerve. 2008;37(2):141-9. [ Links ]
39. Mantegazza R, Beghi E, Pareyson D, Antozzi C, Peluchetti D, Sghirlanzoni A, et al. A multicentre follow-up study of 1152 patients with myasthenia gravis in Italy. J Neurol. 1990;237:339-44. [ Links ]
40. Tapias-Vargas L, Tapias-Vargas LF, Tapias L. Miastenia gravis y el timo: pasado, presente y futuro. Rev Colomb Cir. 2009;24(4):269-82. [ Links ]
41. Richman DP, Agius MA. Treatment of autoimmune myasthenia gravis. Neurology. 2003;61(12):1652-61. [ Links ]
42. Ping- Hung K, Pi-Chuan F. Respiratory Care for Myasthenic Crisis. En: Pruitt JA, editores. A look into Myasthenia Gravis. InTech; 2012. p. 55-75. [ Links ]
43. Werneck LC, Scola RH, Germiniani FM, Comerlato EA, Cunha FM. Myasthenic crisis: report of 24 cases. Arq. Neuropsiquiatr. 2002;60(3-A):519-26. [ Links ]
44. Patra S, Singh AP, Srinivas BC, Manjunath CN. Myasthenia crisis following percutaneous coronary intervention in a patient with late onset myasthenia gravis: successful treatment of a unique case. Cardiovasc Interv Ther. 2013;28(3):279-81. [ Links ]
45. Benatar M, Burns T, Swan AV. Serological, pharmacological and electrophysiological tests for the diagnosis of myasthenia gravis. Cochrane Database of Systematic Reviews. 2010(12):1-10. [ Links ]
46. Heldal AT, Eide GE, Gilhus NE, Romi F. Geographical distribution of a seropositive myasthenia gravis population. Muscle Nerve. 2012;45(6):815-9. [ Links ]
47. Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8(5):475-90. [ Links ]
48. Meriggioli MN, Sanders DB. Muscle autoantibodies in myasthenia gravis: beyond diagnosis? Expert Rev Clin Immunol. 2012;8(5):427-38. [ Links ]
49. Jaretzki A , Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Ann Thorac Surg. 2000;70(1):327-34. [ Links ]
50. Poulas K, Tsibri E, Kokla A, Papanastasiou D, Tsouloufis T, Marinou M, et al. Epidemiology of seropositive myasthenia gravis in Greece. J Neurol Neurosurg Psychiatry. 2001;71(3):352-6. [ Links ]
51. Browning J, Wallace M, Chana J, Booth J. Bedside testing for myasthenia gravis: the ice-test. Emerg Med J. 2011;28(8):709-11. [ Links ]
52. Cherian A, Baheti NN, Iype T. Electrophysiological study in neuromuscular junction disorders. Ann Indian Acad Neurol. 2013;16(1):34-41. [ Links ]
53. Witoonpanich R, Dejthevaporn C, Sriphrapradang A, Pulkes T. Electrophysiological and immunological study in myasthenia gravis: diagnostic sensitivity and correlation. Clin Neurophysiol. 2011;122(9):1873-7. [ Links ]
54. American Association of Electrodiagnostic Medicine. Literature review of the usefulness of repetitive nerve stimulation and single fiber EMG in the electrodiagnostic evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome. Muscle Nerve.2001;24(9):1239-47. [ Links ]
55. Orhan EK, Deymeer F, Oflazer P, Parman Y, Baslo MB. Jitter analysis with concentric needle electrode in the masseter muscle for the diagnosis of generalised myasthenia gravis. Clin Neurophysiology. 2013;124(11):2277-82. [ Links ]
56. Walker MB. Treatment of myasthenia gravis with physostigmine. Lancet. 1934;1:1200-1. [ Links ]
57. Mehndiratta MM, Pandey S, Kuntzer T. Acetylcholinesterase inhibitor treatment for myasthenia gravis. Cochrane Database Syst Rev. 2011;2:CD006986. [ Links ]
58. Szczeklik W, Mitka I, Nowak I, Seczyńska B, Sega A, Węgrzyn W, et al. Plasmapheresis in intensive therapy units. Anestezjol Intens Ter. 2010;42(2):100-6. [ Links ]
59. Saeteng S, Tantraworasin A, Siwachat S, Lertprasertsuke N, Euathringchit J, Wannasopha Y. Preoperative plasmapheresis for elective thymectomy in myasthenia patient: is it necessary? ISRN Neurol. 2013; 2013:1-6. [ Links ]
60. Gajdos P, Chevret S, Toyka KV. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. 2012;12. [ Links ]
61. Brannagan TH, Nagle KJ, Lange DJ, Rowland LP. Complications of intravenous immune globulin treatment in neurologic disease. Neurology. 1996;47(3):674-7. [ Links ]
62. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15(3):291-8. [ Links ]
63. Evoli A, Batocchi AP, Palmisani MT, Lo Monaco M, Tonali P. Long-term results of corticosteroid therapy in patients with myasthenia gravis. Eur Neurol. 1992;32(1):37-43. [ Links ]
64. Bhanushali MJ, Wuu J, Benatar M. Treatment of ocular symptoms in myasthenia gravis. Neurology. 2008;71(17):1335-41. [ Links ]
65. Kupersmith MJ, Moster M, Bhuiyan S, Warren F, Weinberg H. Beneficial effects of corticosteroids on ocular myasthenia gravis. Arch Neurol. 1996;53(8):802-4. [ Links ]
66. Ciafaloni E, Massey JM, Tucker-Lipscomb B, Sanders DB. Mycophenolate mofetil for myasthenia gravis: an open-label pilot study. Neurology. 2001;56(1):97-9. [ Links ]
67. Spring PJ, Spies JM. Myasthenia gravis: options and timing of immunomodulatory treatment. BioDrugs. 2001;15(3):173-83. [ Links ]
68. Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant. 2006;6(5 Pt 1):859-66. [ Links ]
69. Spillane J, Hayward M, Hirsch NP, Taylor C, Kullmann DM, Howard RS. Thymectomy: role in the treatment of myasthenia gravis. J Neurol. 2013;260(7):1798-801. [ Links ]
70. Masaoka A, Yamakawa Y, Niwa H, Fukai I, Kondo S, Kobayashi M, et al. Extended thymectomy for myasthenia gravis patients: a 20 year review. Ann Thorac Surg. 1996;62(3):853-9. [ Links ]
71. Bachmann K, Burkhardt D, Schreiter I, Kaifi J, Schurr P, Busch C, et al. Thymectomy is more effective than conservative treatment for myasthenia gravis regarding outcome and clinical improvement. Surgery. 2009;145(4):392-8. [ Links ]
72. Christison-Lagay E, Dharia B, Vajsar J, Kim PC. Efficacy and safety of thoracoscopic thymectomy in the treatment of juvenile myasthenia gravis. Pediatr Surg Int. 2013;29(6):583-6. [ Links ]
73. Lashkarizadeh MR, Ajami R, Vahedian M, Pourseyedi B, Zeynali H, Fekri MS, et al. Video-Assisted Thoracoscopic Thymectomy as an Optimal Treatment in Myasthenia Gravis. J Minim Invasive Surg Sci. 2012;1(4):144-8. [ Links ]
74. Manlulu A, Lee T, Wan I, Law CY, Chang C, Garzon JC, et al. Videoassisted thoracic surgery thymectomy for nonthymomatous myasthenia gravis. Chest. 2005;128(5):3454-60. [ Links ]
75. Chicaiza-Becerra LA, Garcia-Molina M, Gamboa O, Castañeda- Orjuela C. The cost-effectiveness of open or thoracoscopic thymectomy compared to medical treatment in managing Myasthenia gravis without thymomas. Rev Salud Pública (Bogota). 2012;14(2):260-70. [ Links ]
76. Musthafa CP, Moosa A, Chandrashekharan PA, Nandakumar R, Narayanan AV, Balakrishnan V. Intestinal pseudo-obstruction as initial presentation of thymoma. Indian J Gastroenterol. 2006;25(5):264-5. [ Links ]
77. Akoum R, Brihi E, Chammas S, Abigerges D. Results of radiation therapy for thymoma based on a review of 27 patients. Mol Immunol. 2003;39(17-18):1115-9. [ Links ]
78. Lococo F, Cesario A, Margaritora S, Granone P. Twenty-oneyear survival in an invasive thymoma successfully treated with seven-fold iterative surgery. Interact Cardiovasc Thorac Surg. 2010;11(3):322-4. [ Links ]
79. Awad WI, Symmans P, Dussek JE. Recurrence of stage I thymoma 32 years after total excision. Ann Thorac Surg. 1998;66(6):2106-8. [ Links ]
80. Giaccone G, Wilmink H, Paul MA, van der Valk P. Systemic treatment of malignant thymoma: a decade experience at a single institution. Am J Clin Oncol. 2006;29(4):336-44. [ Links ]
81.Sugie C, Shibamoto Y, Ikeya-Hashizume C, Ogino H, Ayakawa S, Tomita N, et al. Invasive thymoma: postoperative mediastinal irradiation, and low-dose entire hemithorax irradiation in patients with pleural dissemination. J Thorac Oncol. 2008;3(1):75-81. [ Links ]
