










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.27 no.1 Bicaramanga Jan./Apr. 2014
Mecanismos fisiopatológicos del síndrome
antifosfolípidos
Yeison Santamaría-Alza*
*Estudiante de XI nivel de Medicina. Escuela de Medicina. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
Correspondencia: Sr. Yeison Santamaría. Calle 7ª No. 42 - 44 Floridablanca. Santander. Colombia. Correo electrónico: yeison-1807@hotmail.es.
Artículo recibido el 1 de noviembre de 2013 y aceptado para publicación el 16 de diciembre de 2013.
RESUMEN
Introducción: el síndrome antifosfolípidos es una enfermedad de reciente aparición, por lo que no se cuenta con una prevalencia real, sin embargo afecta aproximadamente a 0,5% de la población, principalmente a mujeres; siendo una importante causa de trombosis arterial, venosa o de pequeños vasos y alteraciones obstétricas. Objetivo: hacer una revisión para integrar el conocimiento de los mecanismos implicados en las vías patológicas del síndrome antifosfolípidos que involucran anticuerpos y sustancias endógenas, para que sea usado en el diagnóstico y tratamiento de esta enfermedad. Conclusiones: en el síndrome antifosfolípidos participan varios componentes, entre ellos el anticuerpo anti-Beta2-Glicoproteína1, anticoagulante lúpico, anticuerpos anti-cardiolipina, anticuerpos anti-serin proteasas, anticuerpos anti-anexina A5, sistema del complemento y factor tisular, que al realizar su acción son los causantes de las manifestaciones trombóticas y alteraciones obstétricas. El entender los mecanismos de acción de dichos componentes, puede contribuir al mejoramiento diagnóstico y creación de herramientas terapéuticas para disminuir la mortalidad y morbilidad causada por el síndrome antifosfolípidos. (MÉD.UIS. 2014;27(1):43-50).
Palabras Clave: Anticuerpos Antifosfolípidos. Trombosis. Anticoagulante Lúpico. Anti-cardiolipina.
Phatophysiological Mechanism of the Antiphospholipid Syndrome
ABSTRACT
Introduction: the antiphospholipid syndrome is an emerging disease, so there is no real prevalence, however affects approximately 0.5% of the population, mainly women , being a major cause of arterial , venous, or small vessel and obstetric disorders. Objective: to revise to integrate knowledge of the mechanisms involved in pathological pathways involving antiphospholipid antibody syndrome and endogenous substances, to be used in the diagnosis and treatment of this disease. Conclusions: antiphospholipid syndrome involves several components, including the anti - Beta2 - Glicoproteina 1, lupus anticoagulant, anticardiolipin antibodies, anti- serine proteases, antiannexin A5 antibodies, complement system and tissue factor, that doing their actions are causing thrombotic manifestations and obstetric disorders. Understanding the mechanisms of action of these components can contribute to improved diagnostic and therapeutic tool created to reduce mortality and morbidity caused by antiphospholipid syndrome. (MÉD.UIS. 2014;27(1):43-50).
Keywords: Antiphospholipid Antibodies. Thrombosis. Lupus Anticoagulant. Anti-cardiolipin.
¿Cómo citar este artículo?: Santamaría-Alza Y. Mecanismos fisiopatológicos del síndrome antifosfolípidos.
MÉD.UIS. 2014;27(1):43-50.
INTRODUCCIÓN
El Síndrome Antifosfolípidos (SAF) fue propuesto en el año 1983 por el reumatólogo Graham Hughes1, quien mostró la relación entre trombosis, aborto, enfermedades cerebrales y anticoagulante lúpico2.
La prevalencia real del SAF en la población general se desconoce, pero se calcula que se encuentra alrededor del 0,5%3; sin embargo, entre el 2 y 5% de la población puede tener títulos detectables de los anticuerpos, sin llegar a presentar manifestaciones de la enfermedad4. Por otro lado, el SAF afecta principalmente a las mujeres dando una relación de un hombre por cada cinco mujeres que la padecen. A su vez, se manifiesta principalmente en la tercera década de la vida y solo el 12% de los afectados son mayores de 50 años, de los cuales la gran mayoría corresponden al sexo masculino5. Del total de pacientes con SAF aproximadamente el 50% padecen otra enfermedad autoinmune entre las que se encuentran el lupus eritematoso sistémico, artritis reumatoide y esclerodermia, principalmente4.
En cuanto a las complicaciones generales se ha encontrado que uno de cada cinco pacientes con enfermedad cerebrovascular menor de 50 años padece el SAF6,7 de la misma manera que el 24% de los casos de tromboembolismo (trombosis venosa profunda o tromboembolismo pulmonar) son debidos al SAF8. Respecto a las manifestaciones obstétricas se ha demostrado que entre el 10 al 15% de los abortos9, 15 a 29% de los casos de preeclampsia10 y 25% de las restricciones del crecimiento intrauterino11 son consecuencia del SAF.
La fisiopatológica del SAF es compleja y está caracterizada por un estado de hipercoagulabilidad manifestada por trombosis arterial, venosa y de pequeños vasos, acompañada de alteraciones en el embarazo. Para su diagnóstico es esencial la presencia de al menos un criterio clínico y un criterio de laboratorio12-13. Los criterios diagnósticos clínicos son: trombosis vascular que puede ser arterial, venosa o de pequeños vasos, una o más alteraciones en el embarazo como la muerte fetal inexplicable de morfología normal más allá de la semana 10 de gestación, uno o más partos prematuros con neonatos de características normales antes de la semana 34 de gestación a causa de eclampsia, preeclampsia severa o insuficiencia placentaria, o tres o más abortos espontáneos inexplicables consecutivos antes de la semana 10 de gestación, con exclusión de causas cromosómicas de ambos padres y de causas anatómicas y hormonales maternas. Los criterios de laboratorio son anticoagulante lúpico presente en plasma en dos o más ocasiones con al menos 12 semanas de separación, anticuerpo anticardiolipina tipo IgM o IgG en suero o plasma en títulos medios o altos, en dos o más ocasiones con al menos 12 semanas de separación o anticuerpo Anti-β2 glicoproteína 1 tipo IgM o IgG en suero o plasma, en título superior al percentil 99, en dos o más ocasiones con al menos 12 semanas de separación14.
El mecanismo de acción de los anticuerpos antifosfolípidos no se ha dilucidado completamente. Sin embargo, una de las teorías más aceptadas hasta el momento dice que los anticuerpos se dirigen a los fosfolípidos cargados negativamente, localizados en las membranas celulares de cierto tipo de células como células endoteliales, monocitos y plaquetas en momentos claves del ciclo celular como en la apoptosis o en la activación, cuando se habla de plaquetas. Luego, por medio de receptores se hace una vía de señalización que culmina con la expresión de moléculas de adhesión que a su vez aumentan la adhesión celular y formación de factor tisular que lleva al paciente a un estado protrombótico15.
El objetivo de esta revisión es integrar el conocimiento de los mecanismos implicados en las vías patológicas del SAF que involucran anticuerpos y sustancias endógenas, para que sea usado en el diagnóstico y tratamiento de esta enfermedad, ya que es de gran importancia conocer la fisiopatología de la enfermedad para reconocer, diagnosticar oportunamente y saber actuar frente a las complicaciones del SAF.
Para tal fin, la búsqueda de literatura científica se realizó mediante el uso de las bases de datos PubMed y SciELO, usando las palabras claves: anticuerpos antifosfolípidos, síndrome antifosfolípidos, anti- Beta2-Glicoproteína-1, anticoagulante lúpico y anticardiolipina; encontrando un total de 455 artículos, utilizando como filtros los artículos publicados en los últimos diez años y estudios realizados en humanos, de los que se seleccionaron 34 teniendo en cuenta la importancia de los mismos para los fines de la presente revisión.
ANTICUERPO ANTI-BETA 2-GLICOPROTEINA1
La Beta2-Glicoproteína1 (β2GP1) es una proteína de cadena única altamente glicosilada conformada por 326 aminoácidos16, que se une con avidez a fosfolípidos de carga negativa como la cardiolipina, fosfatidilserina y fosfatidilinositol. Hasta ahora no tiene una función conocida, incluso las personas con deficiencia de esta glicoproteína no presentan ninguna manifestación clínica13.
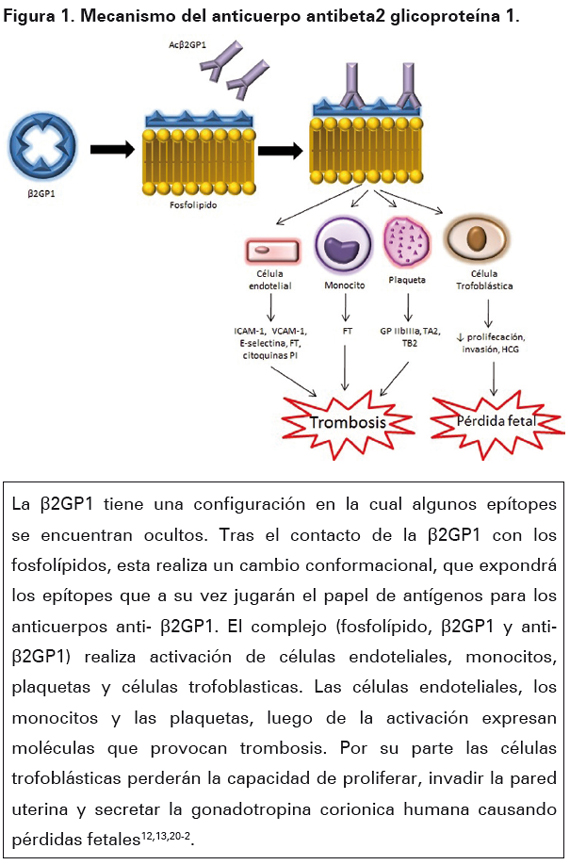
Una vez la proteína se ha unido a los fosfolípidos, la β2GP1 hace cambios conformacionales en su estructura, lo cual culmina con la exposición de epitopes ocultos; en condiciones fisiológicas esta exposición de epítopes es transitoria y no trae ninguna manifestación en los seres humanos. En el momento en el que los epítopes se hallan expuestos es cuando los Anticuerpos anti-β2GP1 (Acβ2GP1) entran en acción, debido a la alta afinidad existente entre los anticuerpos y los epítopes expuestos17,18.
La unión de estos dos componentes hacen que estos epítopes se expresen prolongadamente causando un cambio conformacional permanente en la proteína. La unión de estos tres componentes: β2GP1, fosfolípido y anticuerpo, forman un complejo que se encuentra disponible para interactuar en diferentes reacciones homeostáticas y en diversos receptores celulares16,19.
Reacciones homeostáticas
Dentro de las reacciones homeostáticas que se alteran a causa de este complejo están las vías anticoagulantes y fibrinolíticas. En las vías anticoagulantes alteradas tenemos la inhibición de la vía de la proteína C, inhibición de la actividad de la proteina S e inhibición de la vía de la antitrombina. De las acciones que alteran la fibrinólisis cabe resaltar el aumento de los niveles plasmáticos del inhibidor del activador del plasminógeno y el antígeno del activador tisular del plasminógeno18,19.
Reacciones celulares
Respecto a las células endoteliales, el complejo se une al receptor Anexina A2 que es un potente receptor fibrinolítico. El receptor es una proteína de membrana obligado sin dominio transmembrana por lo que no puede transmitir señales a través de la membrana celular; para que se lleve a cabo la unión es necesaria la existencia del anticuerpo en el complejo. De la misma manera se ha podido confirmar la unión del complejo al receptor tipo Toll 4 que lleva a la iniciación de su vía de señalización y a la activación de las células endoteliales que culmina con un estado protrombótico generado por la expresión de Moléculas de Adhesión Intracelular (ICAM-1), Moléculas de Adhesión Vascular-Celular 1 (VCAM- 1), E-selectina, aumento en la producción de factor tisular e incremento en la producción y secreción de citoquinas proinflamatorias16,18,19.
Así mismo, el complejo Ac-β2GP1 β2GP1, activa a los monocitos a través de los mismos receptores por los que activa las células endoteliales: Anexina A2 y receptor tipo Toll 4. La activación de los monocitos culmina con un "up regulation" del factor tisular aumentando de esta manera sus niveles plasmáticos16,18,19.
Las plaquetas son activadas por el complejo antígeno-anticuerpo a través de básicamente dos receptores: receptor de lipoproteína de baja densidad relacionado con la proteína 8 y el receptor adhesivo plaquetario de la glicoproteína Ibα. La activación de las plaquetas lleva a una vía de señalización celular que termina con aumento en la expresión de glicoproteína IIb-IIIa, aumento en la síntesis de tromboxano A2 e inducción de la producción de tromboxano B2 a través de proteinquinasas activadas por mitógeno P38 con la consecuente fosforilación de la fosfolipasa A2 citosólica plaquetaria18,19.
En la actualidad se ha podido demostrar la expresión de β2GP1 en las membranas celulares de las células trofoblásticas; factor que podría explicar el especial trofismo hacia la placenta que presentan las pacientes con SAF. La acción del complejo antígeno-anticuerpo impide algunas funciones de estas células como la proliferación, la invasión y la secreción de la gonadotropina coriónica humana; por otra parte estimula la apoptosis celular. Sumando esos dos factores, la existencia del complejo causa en la placenta trombosis e infarto18,19. Todo lo anterior explica las manifestaciones vasculares y las complicaciones obstétricas del SAF (Ver Figura 1)12, 13, 20-2.
ANTICOA GULANTE LÚPICO
Es una inmunoglobulina tipo IgG, IgA o una mezcla de las dos que interfiere con las reacciones de coagulación dependientes de fosfolípidos. Los anticuerpos no están dirigidos contra los factores de coagulación sino contra ciertos epítopes de fosfolípidos aniónicos. Se puede encontrar aumentado en diferentes entidades clínicas como enfermedades autoinmunes, exposición a drogas y antibióticos, infecciones bacterianas, virales o protozoarias y desordenes linfoproliferativos. Aunque el Anticoagulante Lúpido (AL) en el laboratorio prolonga las pruebas de coagulación no se correlaciona con procesos hemorrágicos, por el contrario se asocia a procesos trombóticos23.
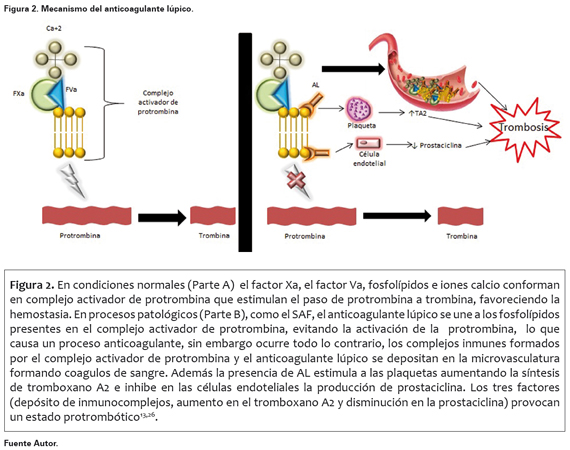
En condiciones normales, en la cascada de la coagulación se forma el complejo activador de la protrombina, del cual hacen parte el factor Xa, Va, iones calcio y fosfolípidos de origen tisular o plaquetario24. Este complejo se encarga de la iniciación de la trombina que activará el fibrinógeno liberando monómero de fibrina. En presencia del AL, este se une a los fosfolípidos del complejo activador de la protrombina, dejándolo parcialmente funcionante, comportándose como un inhibidor adquirido de la coagulación llevando al individuo a una prolongación de la coagulación dependiente de fosfolípidos. Posteriormente estos complejos se depositan en el lecho vascular lo que produce los eventos trombóticos23,25.
Adicional a lo anterior, la presencia de AL se ha asociado a la disminución en la síntesis de prostaciclina en las células endoteliales acompañado de aumento en la producción del tromboxano A2 plaquetario. Ambos factores favorecen la aparición de manifestaciones trombóticas26.
El anticuerpo antifosfolípido que mayor información brinda respecto a las características clínicas del SAF es el AL (Ver Figura 2)13.
ANTICUERPOS ANTI-CARDIOLIPINA
La cardiolipina es un lípido aniónico, componente de las membranas celulares, también es componente de las lipoproteínas. Está compuesta por una unidad dimérica formada por cuatro ácidos grasos con dos cargas negativas. Juega un papel importante en la mitocondria al interactuar en el proceso de la fosforilación oxidativa27.
El Anticuerpo Anti-Cardiolipina (aCL) es un autoanticuerpo dirigido contra los complejos fosfolipidos-proteínas de las membranas celulares, generalmente son de la clase IgG. Para su unión, estos anticuerpos requieren la presencia de β2GP1. Dentro de los mecanismos fisiopatológicos propuestos para el anticuerpo aCL se encuentran: interferencia con la producción endotelial de prostaciclina, interferencia de la vía de la trombomodulina ya sea en la modulación de la proteína C o en la amplificación de la respuesta mediada por la proteína S, interferencia en la función de la antitrombina, promoción de la activación plaquetaria con la interacción mediada por fosfolípidos o por interferencia en el paso de prekalikreína a kalikreína28.
Este anticuerpo ha sido ampliamente relacionado con las manifestaciones obstétricas, ya que un evento clásico encontrado en las mujeres con títulos elevados de aCL es el infarto placentario extenso. El mecanismo por el cual los anticuerpos aCL afectan la placenta es por medio de la inhibición de la proliferación del trofoblasto lo que evita la invasión de las arterias espirales maternas, y por tal motivo el flujo sanguíneo no se modifica para soportar las necesidades básicas de la gestación27.
Para que se presenten manifestaciones es necesaria la existencia de títulos medios o altos. Además de las manifestaciones obstétricas, el anticuerpo aCL se ha relacionado con manifestaciones nueropsiquiátricas29.
ANTICUERPO ANTISERIN PROTEASAS
ANTICUERPOS ANTITROMBINA
La trombina es la enzima encargada de la degradación del fibrinógeno a monómeros de fibrina. Esta enzima es regulada por la antitrombina mediante la fijación de la trombina a la superficie de las células endoteliales. En el SAF los anticuerpos se unen a la trombina en el lugar que fisiológicamente lo hace la antitrombina que provoca una excesiva activación de la enzima, que a su vez culmina con la estabilización de los coágulos a causa del aumento en la concentración de fibrina21.
Anticuerpos anti-Proteína C
La proteína C es una glicoproteína transmembrana vitamina K dependiente, que actúa como anticoagulante natural en su forma activa. La activación se da cuando el complejo trombinatrombomodulina se une a la proteína C. Su función, cuando se encuentra activada, es llevar a cabo la degradación del factor Va y VIIIa de la coagulación, lo que termina con la inhibición en la formación de trombina, que evita la formación de coágulos21.
La proteína S, otra proteína vitamina K dependiente se encarga de la amplificación de la actividad de la proteína C activada, por medio de la formación de un complejo Proteína C activada - Proteína S sobre las superficies de fosfolípidos21,28.
En el SAF la función de la proteína C se ve truncada por la unión de anticuerpos a la proteína C evitando su activación, anticuerpos dirigidos contra la Proteína C activada que disminuyen la amplificación por medio de la proteína S, anticuerpos contra la proteína S o anticuerpos contra la trombomodulina. Los anteriores mecanismos causan la eliminación o disminución de la actividad anticoagulante de la proteína C28.
La acción de los anticuerpos sobre la proteína C se relaciona con las manifestaciones relacionadas con la trombosis venosa; sin embargo no se ha vinculado con la trombosis arterial o las complicaciones obstétricas12,21.
Anticuerpos antiActivador Tisular del plasminógeno
El activador tisular del plasminógeno es una proteína perteneciente a las serín proteasas que se encuentra en las células endoteliales encargada de catalizar la conversión de plasminógeno a plasmina21.
Los anticuerpos en el SAF se unen al activador tisular del plasminógeno e impiden la conversión de plasminógeno en plasmina con la consecuente disminución en la concentración del producto de la reacción y aumento en la estancia de los coágulos en circulación21.
Anticuerpos antiplasmina
La plasmina es la proteína resultado de la activación del plasminógeno, tiene como finalidad lograr la degradación de la fibrina, disolviendo así los coágulos y mejorando el flujo lineal de la sangre. En el SAF, algunos anticuerpos del tipo Inmunoglobulina G se unen a la plasmina y de esta manera se entorpece la función de la proteína prolongando la vida media de los coágulos de fibrina aumentando el riesgo de trombosis21.
Anticuerpos antiAnexina A5
La anexina A5 es una proteína celular cuya función no ha sido esclarecida, sin embargo, por su alta afinidad para unirse a los fosfolípidos con carga negativa y su capacidad para realizar inhibición de reacciones de coagulación dependiente de fosfolípidos, se le ha relacionado con una potente función de anticoagulante fisiológico. Los anticuerpos dirigidos contra la anexina A5, relacionados con el SAF, hacen interrupción en la función de esta que termina con el aumento en la producción de trombina, lo que podría explicar la trombosis placentaria y la pérdida fetal presente en el SAF13,15,20.
Se ha hecho un especial vínculo entre los anticuerpos mencionados y la pérdida gestacional debido a que en condiciones fisiológicas se han encontrado altos niveles de anexina A5 en el trofoblasto por lo que podría estar implicada en la fusión de las células, proliferación de las membranas y la formación del sincitio14,30.
En recientes estudios se ha demostrado la disminución de la anexina A5 en la placenta de pacientes con pérdidas gestacionales que presentan el SAF, comparada con la concentración encontrada en la placenta de aquellas mujeres que teniendo abortos no presentan el SAF31. Adicionalmente, se ha podido comprobar que solo los altos títulos de anticuerpos IgG contra anexina A5 se han relacionado con las manifestaciones clínicas propias del SAF, ya que con títulos bajos no se presenta ninguna manifestación relevante. De la misma manera no se ha logrado vincular a los anticuerpos IgM con las manifestaciones trombóticas30,32.
SISTEMA DEL COMPLEMENTO
Activación de células endoteliales
Luego de la activación del complemento se generan porciones activas del mismo que tienen la capacidad de unirse y al mismo tiempo activar a células inflamatorias y a células endoteliales. Esta activación se puede llevar a cabo por medio de C5b-C9 (complejo de ataque de membrana) o a través de los efectos dados por la unión de C5a a su receptor ubicado en las membranas de dichas células. La activación de las células endoteliales concluye con un aumento en la producción de factor tisular29.
Daño del trofoblasto
En la placenta, el complejo β2GP1-Ac se encarga de la activación del complemento, llevándolo a la vía clásica y generando porciones activas como C5a y C3a. Estas porciones con actividad se encargan de llevar a cabo la activación de neutrófilos, monocitos y plaquetas que van a producir mediadores inflamatorios como radicales de oxígeno, factor tisular, enzimas proteolíticas y citoquinas como el TNFα. Los mediadores inflamatorios inician su actividad y llevan a un daño variable en el trofoblasto, que a largo plazo podría causar restricción del crecimiento intrauterino, preeclamsia o pérdida fetal19,29,31.
FACTOR TISULAR
El Factor Tisular (FT) es una glicoproteína transmembrana de cadena simple compuesta por 263 residuos de aminoácidos, que funciona como receptor para el factor VIIa de la coagulación que se encuentra expuesta en los fibroblastos de las paredes de los vasos sanguíneos. El FT actúa como iniciador de la coagulación. Una vez se ha unido el factor VII activado (FVIIa) al factor tisular (FT) se forma un complejo que tiene una alta afinidad por el Factor X (FX). Cuando el FX se une al complejo FT-FVIIa, por actividad proteolítica, se convierte en Factor X activado (FXa). Finalmente, el FXa se une con el factor V activado en las membranas con carga negativa promoviendo la actividad de la protrombinasa y convirtiendo la protrombina en trombina. Esta última realiza el paso de fibrinógeno a monómeros de fibrina. No obstante, cuando existe alguna lesión vascular o cuando existen algunos estímulos químicos como lipopolisacáridos bacterianos, factor de necrosis tumoral α, interleucina 1 o complejos inmunes, esta glicoproteína se expresa en las células endoteliales y en los monocitos, facilitando la unión del FT con el FVIIa, encargándose de realizar la activación de la cascada de la coagulación y finalizando con la formación de trombos que mejorarán la homeostasia del lugar de injuria vascular21,29,33.
El FT no solo inicia la coagulación por la vía anteriormente descrita, también la puede iniciar por medio de la iniciación de una vía de señalización que incluye la activación de una proteína G acoplada a receptores con actividad proteolítica: PAR1 y PAR2, de mayor significancia fisiopatológica el receptor PAR2. Posterior a la unión del FT y el receptor PAR2 se inicia la coagulación sanguínea que simula la función inflamatoria de las células del sistema inmune31.
Si bien es cierto que la expresión del TF es necesaria para mantener la hemostasia propia del cuerpo, su excesiva expresión, como la que se encuentra en el SAF hace que los pacientes que padecen este síndrome manifiesten trombosis34.
La cantidad del complejo FT-FVII-FX depende de la cantidad de factor tisular que se genere y por lo tanto a mayor concentración de FT mayores serán las manifestaciones de la coagulación. En el SAF la expresión de los anticuerpos aumenta la producción de FT por parte de las células endoteliales y los monocitos lo que conlleva a una activación excesiva de la cascada de coagulación y presentando en el paciente múltiples manifestaciones trombóticas33.
CONCLUSIONES
En el presente trabajo se constató que la prevalencia del SAF es desconocida, esto debido a que es una entidad "joven" y que hasta el momento, no se encuentran reportados estudios epidemiológicos que revelen esta información. Sin embargo, es recomendable sospechar la presencia de esta entidad en pacientes con antecedentes trombóticos de corta edad y mujeres con múltiples alteraciones obstétricas.
Entre los anticuerpos que más se han relacionado con el SAF se encuentran el anti-B2GP1, anticoagulante lúpico y anticuerpos anticardiolipina. No obstante, además de estos, se ha encontrado relación con anti-serin proteasas, anticuerpos anti-anexina A5 y sustancias endógenas como: factor tisular y sistema del complemento, en las manifestaciones clínicas vistas en los pacientes con SAF.
Se hace necesario realizar estudios donde se establezca la prevalencia real, tanto local como nacional y de ahí tener un punto de partida para posteriores estudios que ayuden a mejorar el diagnóstico, tratamiento y pronóstico de la enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1. Gray JM, Khamashta MA. Antiphospholipid syndrome: coming of age.Reumatol Clin.2011;7(3):151-3. [ Links ]
2. Hughes GR. Thrombosis, abortion, cerebral disease and the lupus anticoagulant.Br Med J (Clin Res Ed).1983 October 15;287(6399):1088-9. [ Links ]
3. Mok CC, Tang SS, To CH, Petri M. Incidence and risk factors of thromboembolism in systemic lupus erythematosus: a comparison of three ethnic groups.Arthritis Rheum.2005 Sep;52(9):2774-82. [ Links ]
4. Horstman LL, Wenche JY, Bidot CJ, Ahnn YS, Kelley RE, Zivadinov R, et al. Antiphospholipid antibodies: paradigm in transition.J Neuroinflammation.2009 Jan 20;6:3. [ Links ]
5. Mulla MJ, Brosens JJ, Chamley LW, Giles I, Pericleous C, Rahman A, et al. Antiphospholipid antibodies induce a pro-inflammatory response in first trimester trophoblast via the TLR4/MyD88 pathway.Am J Reprod Immunol.2009 Aug;62(2):96-111. [ Links ]
6. Ruiz-Irastorza G, Egurbide MV, Ugalde J, Aguirre C. High impact of antiphospholipid syndrome on irreversible organ damage and survival of patients with systemic lupus erythematosus.Arch Intern Med.2004;164(1):77-82. [ Links ]
7. Koniari I, Siminelakis SN, Baikoussis NG, Papadopoulos G, Goudevenos J, Apostolakis E. Antiphospholipid syndrome; its implication in cardiovascular diseases: a review.J Cardiothorac Surg.2010 Nov 3;5:101 [ Links ]
8. Roldan V, Lecumberri R, Muñoz-Torrero JF, Vicente V, Rocha E, Brenner B, et al. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry.Thromb Res.2009;124(2):174-7. [ Links ]
9. Silver RM. Fetal death.Obstet Gynecol.2007;109(1):153-67. [ Links ]
10. Gómez JM, Aguirre N, Quintero A, Gómez J. Preeclampsia y anticuerpos antifosfolípidos.Revista Colombiana de Obstetricia y Ginecología.2000 Jul;51(3):1-5. [ Links ]
11. Smith GC, Crossley JA, Aitken DA, Pell JP, Cameron AD, Connor JM, et al. First-trimester placentation and the risk of antepartum stillbirth.JAMA.2004;292(18):2249-54. [ Links ]
12. Tripodi A, de Groot PG, Pengo V. Antiphospholipid syndrome: laboratory detection, mechanisms of action and treatment.J Intern Med.2011 Aug;270(2):110-22. [ Links ]
13. Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome.The Lancet.2010 Oct;376(9751):1498-509. [ Links ]
14. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006 Feb;4(2):295-306. [ Links ]
15. Lim W. Antiphospholipd antibody syndrome.Hematology Am Soc Hematol Educ Program.2009:233-9. [ Links ]
16. Núñez-Álvarez CA, Cabiedes J. Pathogenic mechanisms of the anti-phospholipid antibodies. Reumatol Clín.2011 Jan - Feb;7(1):72-6. [ Links ]
17. Levine JS, Branch DW, Rauch J. The antiphospholipid Syndrome.N Engl J Med.2002 Mar 7;346:752-63. [ Links ]
18. Espinosa G, Cervera R. Antiphospholipid syndrome.Arthritis Research and therapy.2008;10(6):230. [ Links ]
19. Gropp K, Weber N, Reuter M, el al. B2-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood.2011 Sep 8;118(10):2774-83. [ Links ]
20. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nature reviews Rheumatology.2011 Jun;7(6):330-9. [ Links ]
21. Chen PP, Giles I. Antibodies to serin proteases in the antiphospholid syndrome. Current Rheumatology Reports. 2010 Feb;12(1): 45-52. [ Links ]
22. Oku K, Amengual O, Atsumi T. Pathophysiology of thrombosis and pregnancy morbidity in the antiphospholipd syndrome. Eur J Clin Invest.2012 Oct;42(10):1126-35. [ Links ]
23. Razo D. Síndrome antifosfolipidos y anticoagulante lúpico, anticuerpos antifosfolípidos. Revista Mexicana de Patología Clínica. 2000 Jul-Sep;47(3):168-71. [ Links ]
24. Galli M, Luciani D, Bertolini G, Barbui T. Anti-ß2-Glycoprotein I, antiprothrombin antibodies, and the risk of thrombosis in the antiphospholipid syndrome. Blood Journal American Society of Hematology.2003;102:2717-23. [ Links ]
25. Galindo CG, Bernardez FJ, Hernández I, Ayala AR. Síndrome antifosfolipídico y reproducción humana. Ginecol Obstet Mex.2007;75(5):277-85. [ Links ]
26. Montero PJ, Franco V, Strauss S, de la Peña A. The antiphospholipid syndrome. Revista de Medicina Interna y Crítica.2008;5(1):27-40 [ Links ]
27. Chamley LW, Duncalf AM, Mitchell MD, Jhonson PM. Action of anticardiolipin and antibodies to ß2-Glycoprotein-I on trophoblast proliferation as a mechanism for fetal death.The Lancet.1998 Sep 26;352(9133):1037-8. [ Links ]
28. Brick RL. Hematological complications in obstetrics, pregnancy and gynecology. 1st Ed. New York: Cambridge University Press; 2006 Pp. 196-9. [ Links ]
29. Espinosa G, Cervera R, Front J, Shoenfeld Y. Mechanisms of thrombosis in the antiphospholid syndrome. Inmunología.2003;22(1):53-62. [ Links ]
30. de Laat B, Derksen RH, Mackie IJ, Van Heerde Wl. Annexin A5 polymorphism and the presence of anti-annexin A5 antibodies in the antiphospholipid syndrome.Ann Rheum Dis.2006 Nov;65(11):1468-72 . [ Links ]
31. Di Prima FA, Valenti O, Hyseni E, Giorgio E, Faraci M, Renda E, et al. Antiphospholipid Syndrome during pregnancy: the state of the art. J Prenat Med.2011;5(2):41-53. [ Links ]
32. Ueki H, Mizushina T, Laoharatchatathanin T, Terashima R, Nishimura Y, Rieanrakwong D, et al. Loss of maternal annexin A5 increases the likehood of placental platelet thrombosis and foetal loss.Sci Rep.2012;2:827. [ Links ]
33. Weiler H. Tracing the molecular pathogenesis of antiphospholipid syndrome. The Journal of Clinical Investigation.2008 Oct;118(10):3276-8. [ Links ]
34. Boles J, Mackman N. Role of tissue factor in thrombosis in antiphospholipid antidoby syndrome.Lupus Journal.2010 Apr;19(4):370-8. [ Links ]
