










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.27 no.3 Bicaramanga Sep./Dec. 2014
Síndrome de Reye congénito asociado a varicela
materna
César Augusto Fernández-Dulcey*
Ernesto García Ayala**
Luis Alfonso Pérez***
* Médico. IPS Coomultrasan. Bucaramanga. Santander. Colombia.
** Médico Patólogo. Profesor titular. Departamento de Patología. Universidad Industrial de Santander. Facultad de Salud. Escuela de Medicina. Bucaramanga. Santander Colombia.
*** Médico Pediatra Neonatólogo. Profesor titular de cátedra. Departamento de Pediatría. Universidad Industrial de Santander. Jefe de la Unidad Neonatal. Hospital Universitario de Santander. Bucaramanga. Santander. Colombia.
Correspondencia: Dr. César Fernández-Dulcey. Dirección: Calle 199 # 38-08 Barrio Nogales. Floridablanca. Santander. Colombia.
Correo electrónico: cesarfernandezdulcey@gmail.com
Artículo recibido el 2 de Mayo de 2014 y aceptado para publicación el 8 de Agosto de 2014.
RESUMEN
Introducción: el síndrome de Reye es una encefalopatía aguda asociada a una degeneración grasa del hígado que usualmente es precedida de una infección respiratoria o varicela y tiene una alta prevalencia en niños menores de seis años. Objetivo: reportar un caso clínico de síndrome de Reye congénito asociado a la infección por varicela adquirida de la madre. Presentación de caso: se describen los hallazgos de la autopsia, la respectiva correlación clinicopatológica de un recién nacido de sexo masculino de 37 semanas de gestación, hijo de madre con varicela activa desde cuatro días antes del parto, quien presentó súbitamente palidez generalizada, bradicardia y apnea. Resultados: el examen histopatológico encontró en el citoplasma de los hepatocitos y túbulos renales un compromiso vacuolar que correspondía a grasa. En el cerebro se evidenció severo edema, sin inflamación perivascular o meníngea. Conclusión: corresponde a un caso de síndrome de Reye congénito asociado a varicela materna, que terminó manifestándose clínicamente como muerte súbita. Podría ser la primera publicación de un caso de síndrome de Reye congénito asociado a varicela materna. MÉD.UIS. 2014;27(3):113-121.
Palabras Clave: Síndrome de Reye. Varicela. Embarazo. Recién nacido. Anomalías congénitas.
Congenital Reye syndrome associated to varicella infection in pregnancy
ABSTRACT
Introduction: Reye's syndrome is an acute encephalopathy associated with fatty degeneration of the liver that usually is preceded by a respiratory infection or chickenpox and is highly prevalent in children under 6 years old. Objective: to report a clinical case of congenital Reye's syndrome associated with varicella infection acquired from the mother. Case report: we describe the autopsy findings with the respective clinicopathological correlation of a male newborn of 37 weeks of gestation, son of mother with active varicella from 4 days before birth, who presented sudden paleness, bradycardia and apnea. Results: histopathologic examination found in the cytoplasm of hepatocytes and renal tubules a vacuolar commitment that corresponds to fat. The brain showed severe edema without perivascular or meningeal inflammation. Discussion and conclusion: it corresponds a case of congenital Reye's syndrome associated with varicella infection in pregnancy, who finished clinically as sudden death. This could be the first published case of congenital Reye's syndrome associated with varicella infection in pregnancy. MÉD.UIS. 2014;27(3):113-121.
Keywords: Reye Syndrome. Chickenpox. Pregnancy. Newborn. Congenital abnormalities.
¿Cómo citar este artículo?: Fernández-Dulcey CA, García E, Pérez LA. Síndrome de Reye congénito
asociado a varicela materna: presentación de un caso. MÉD.UIS. 2014;27(3):113-121.
INTRODUCCIÓN
El síndrome de Reye fue descrito por primera vez en 1963 por los patólogos australianos Ralph Reye, Graeme Morgan y Jim Baral1, al cual debe su nombre esta enfermedad, vigente hasta el momento. Se define como una encefalopatía aguda asociada a una degeneración grasa del hígado.
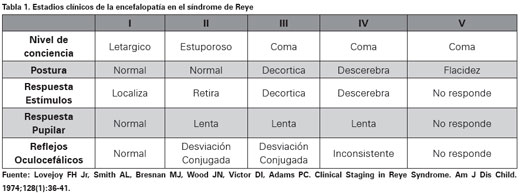
El síndrome de Reye es una enfermedad bifásica que consta de un pródromo viral agudo (infección respiratoria, varicela, gastroenteritis, etc.) que suele mejorar o desaparecer durante tres o cuatro días con tratamiento sintomático. Aparece posteriormente vómito recurrente, que avanza a signos de encefalopatía aguda como letargo, agitación, delirio, estupor progresivo o coma2-5. Frederick Lovejoy describió cinco estadíos clínicos dependiendo del grado de la encefalopatía y el pronóstico empeora a medida que aumenta la afectación neurológica6 (Ver Tabla 1).
Este orden de síntomas no es seguido típicamente en los lactantes, en ellos el síndrome de Reye se puede manifestar con diarrea, sin vómito y se puede acompañar de alteraciones respiratorias como hiperventilación, respiración irregular o apnea. Se debe sospechar cuando se presenta ausencia significativa de fiebre e ictericia durante el periodo de presentación, hepatomegalia, una disfunción hepática mostrada por la elevación de amoniaco o transaminasas de tres a seis veces su valor normal, disminución de los niveles de glucosa sérica, sin documentar ninguna otra causa etiológica aparente. También puede presentarse acidosis, tiempo de protrombina prolongado, lactato deshidrogenasa aumentada o aparecer convulsiones después de un corto periodo de fiebre y vómito. Puede evidenciarse edema cerebral en la tomografía y la punción lumbar puede mostrar un fluido cerebroespinal acelular, sin pleocitosis, con disminución de los niveles de glucosa7-9.
Esta enfermedad presenta una etiología heterogénea predominantemente infecciosa, tóxica o metabólica3,13,14. En la práctica clínica hay dos formas de síndrome de Reye: el síndrome de Reye-like producido por defectos enzimáticos; por ejemplo la deficiencia de acyl-CoA deshidrogenasa de cadena media que causa daño en la beta oxidación de lípidos, o déficit de ornitina-transcarbamilasa que genera hiperamonemia; por otro lado está el llamado síndrome de Reye "idiopático" que se observa en personas predispuestas genéticamente asociado con un factor exógeno que contribuye a la iniciación de este desorden metabólico, como la ingestión de salicilatos, acetaminofén, fenotiazidas, metoclopramida, zidovudina, ácido valproico, didanosina, algunas toxinas como la aflatoxina, bongkrekate, hipoglicina, atractylis gummifera, aceite de margosa, pesticidas, polioxietileno o bien como infecciones virales15,16.
Sin embargo, la causa precisa del síndrome de Reye es desconocida y frecuentemente precedida por una infección viral, usualmente varicela, influenza A, influenza B, varicela zóster, parainfluenza, coxackie, citomegalovirus, Epstein-Barr, dengue, sarampión, virus de la poliomielitis, paramixovirus, picornavirus, reovirus, adenovirus, enterovirus, una gastroenteritis, una infección del tracto respiratorio superior y el uso de ácido acetil salicílico para su tratamiento8,17-24. Los salicilatos fueron detectables en sangre en el 82% de los pacientes8.
El síndrome de Reye es una enfermedad poco frecuente que si no es diagnosticada y tratada a tiempo puede causar la muerte. Es usual la presentación en menores de 18 años y tiene una alta prevalencia en niños menores de 6 años8. Entre 1980 y 1997, 1207 casos de síndrome de Reye en menores de 18 años fueron reportados por el Center of Disease Control (CDC) de los Estados Unidos. El pico de incidencia máximo ocurrió en 1980 con 555 casos reportados. La incidencia fue reducida de 0,6 casos por 100 000 niños en 1979 a menos de 0,1 casos por 100 000 niños en 1989. Entre 1994 y 1997 no se han detectado más de dos pacientes anualmente en Estados Unidos8. Esta significativa disminución de la incidencia no solo se debe a las políticas de salud pública que recomendaron disminuir el uso de salicilatos en niños con fiebre o enfermedades virales, sino también a su difícil diagnóstico y al hecho de no haber pautas claras para su notificación. La incidencia actual es de menos de 0,03 casos por 100 000 individuos menores de 18 años11. No se conoce la incidencia en Colombia pero existe una publicación en 1992 de una mujer de 20 años de edad con síndrome de Reye, entidad que se presenta raramente en adultos12.
Macroscópicamente, el cerebro muestra marcado edema y los cortes histopatológicos con microscopia de luz se destacan por la ausencia de necrosis e inflamación. El hígado es microscópicamente de un color amarillo o blanco, los hepatocitos muestran difusamente una acumulación grasa microvesicular sin evidencia de infiltrado inflamatorio y con presencia de un núcleo central. Los cambios a la microscopia electrónica muestran notable alteración en la morfología mitocondrial con pleomorfismo, ensanchamiento en el tamaño y reducción en el número de ellas. También se destaca la depleción de los niveles de glucógeno10.
La evidencia sugiere que en el síndrome de Reye existe un fracaso mitocondrial como resultado de la inhibición de la fosforilación oxidativa y la oxidación de ácidos grasos de cadena larga, y como consecuencia probable surge la esteatosis microvacuolar. Se ha demostrado en una variedad de células que la acumulación tisular de ácidos grasos de cadena larga inicia la apoptosis por el incremento de las ceramidas17.
En las células normales se han encontrado proteínas de desacople mitocondrial cuyo papel aún es desconocido, las cuales están ausentes en células de pacientes de síndrome de Reye. Su expresión está aumentada en condiciones en las cuales los ácidos grasos de cadena larga se acumulan, lo cual sugiere, que por la estimulación de la respiración mitocondrial estas refuerzan la beta oxidación y protegen contra la apoptosis17.
En el síndrome de Reye relacionado con la toxicidad medicamentosa, la inducción del poro de transición de permeabilidad mitocondrial es sugerido como un mecanismo fisiopatológico que causa la injuria del hígado por el deterioro de la oxidación de la mitocondria25.
El propósito de esta presentación de caso es describir de un recién nacido con antecedente de varicela materna quien presentó muerte súbita y en los hallazgos histopatológicos se evidenció infiltración grasa hepática y edema cerebral que permitió establecer un diagnóstico de síndrome de Reye.
PRESENTACIÓN DEL CASO
Se recibe el 16 de Julio de 2000 un neonato de sexo masculino, producto de tercera gestación de una mujer de 26 años con un embarazo de 37 5/7 semanas y con varicela activa desde cuatro días antes del parto. El parto fue vaginal sin complicaciones, se intentó uteroinhibición la cual fue fallida. El peso fue de 2720 gr y de 51 cm de talla al nacer, con Apgar de 10 al minuto y de 10 a los cinco minutos. Se encontró al recién nacido alerta, con adecuada adaptación neonatal, normocéfalo y fontanela normotensa, con frecuencia cardiaca y respiratoria normales, llenado capilar menor de dos segundos y pulsos distales palpables. Presentó las pupilas isocóricas reactivas a la luz, córnea transparente, reflejo rojo normal, boca sin alteraciones, paladar indemne, cuello móvil, orejas normoimplantadas. A la auscultación se encontraron ruidos cardíacos rítmicos, sin soplos, ruidos respiratorios adecuados, con murmullo vesicular en ambos campos pulmonares y no hubo ruidos sobreagregados. Presentó un Silverman- Anderson de cero. Se encontró un abdomen blando, sin masas, el muñón umbilical con dos arterias y una vena. Se halló el ano permeable, genitales masculinos normoconfigurados y testículos en número de dos en escroto. Al examen de cadera los signos de Barlow y Ortolani fueron negativos. En el examen neurológico se evidenció un niño reactivo, con reflejos palmar, succión y presión positivos.
Se consideró como un recién nacido a término con adecuado peso para la edad gestacional, sin lesiones activas en piel, ni otros signos de infección por varicela. Recibió profilaxis con 1 mg de vitamina K intramuscular, oftálmica y umbilical. Se indicó gammaglobulina específica para varicela de acuerdo a las pautas de la Academia Americana de Pediatría26.
Se instauró aislamiento estricto a la madre por lo que al recién nacido se le administró fórmula de iniciación con tolerancia a la vía oral. El paciente permaneció en buenas condiciones generales, rosado y activo. A las 16 horas posterior al nacimiento se le canalizó vena y se le administró la gammaglobulina específica para varicela (Varitect®), presentación de uso endovenoso y cuya dosis es de 1 cm3/kg mediante la mezcla con 20 cm3 de solución salina normal pasando dos gotas por minuto en mínimo 10 minutos, la infusión duró 40 minutos sin complicaciones. Al terminar la administración del medicamento presentó palidez mucocutánea generalizada, frecuencia cardiaca menor de 30 latidos por minuto, aspecto moteado y apnea. Se inició reanimación neonatal, se procedió a intubar con tubo N° 3,5, se proporcionó apoyo ventilatorio y compresiones torácicas durante un minuto sin aumento en la frecuencia cardiaca ni esfuerzo espontáneo, por lo que se suministró adrenalina 0,1 mg/kg a través del tubo orotraqueal. Se continuaron compresiones torácicas y apoyo ventilatorio sin mejoría. Se canalizó vena umbilical con una sonda No. 8, se pasó un bolo de 30 cm3 de solución salina normal en cinco minutos, hidrocortisona 30 mg endovenosos y adrenalina 0,1 mg/kg cada 5 minutos sin respuesta evidenciada por aumento de la frecuencia cardiaca o inicio de la respiración espontánea. Transcurridos 15 minutos después de iniciada la reanimación avanzada, el paciente presenta una marcada palidez mucocutánea, livideces, pupilas midriáticas no reactivas, asistolia y apnea. Se decide suspender las maniobras de reanimación y el paciente fallece.
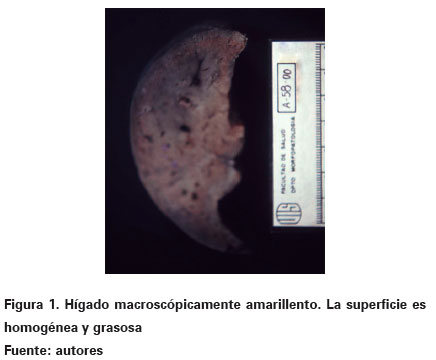
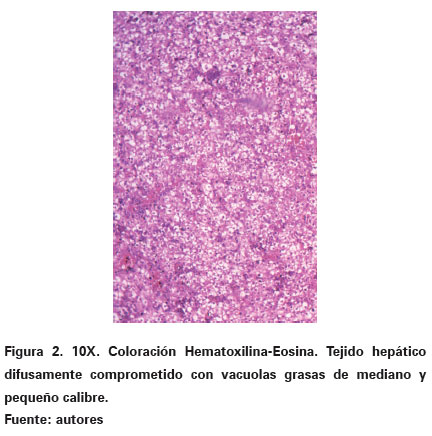
En patología se recibió el cadáver de un recien nacido de sexo masculino el cual pesó 2800 gr y midió 51,5 cm. Al abrir la cavidad abdominal se observaron las vísceras in situ y se destacó como el hallazgo macroscópico más importante la presencia de un hígado aumentado de tamaño, con tonalidad amarillenta y un incremento en un 50% en su peso y volumen (Ver Figura 1). La superficie de corte fue homogénea y difusa con sensación táctil untuosa. En atención a los hallazgos anteriormente descritos, se decidió realizar improntas las cuales fueron coloreadas con rojo oleoso para evidenciar grasa y se obtuvieron extendidos que mostraron hepatocitos positivos para vacuolas de grasa en su citoplasma. Desde el punto de vista histológico se destacaba preservación de la arquitectura normal trabecular y en su mayoría, se visualizaba de una manera difusa y dentro de los citoplasmas de los hepatocitos, un compromiso vacuolar con elementos pequeños e intermedios, bien definidos, claros, prácticamente sin evidencia de material dentro de las mismas (Ver Figura 2). No se observó ningún tipo de necrosis así como tampoco se suscitaba una importante reacción inflamatoria.
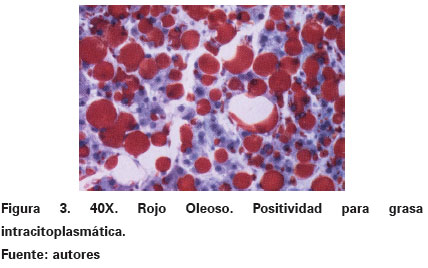
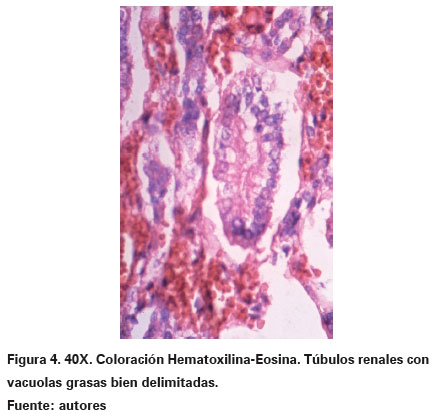
Las coloraciones especiales de histoquímica que fueron realizadas mostraron negatividad para las coloraciones de ácido peryódico de Shiff y mucicarmín. Sin embargo, había positividad para grasa con las técnicas de sudán IV y rojo oleoso, en las cuales se evidenció que las vacuolas tomaron una coloración roja intensa (Ver Figura 3). Los riñones eran simétricos, con lobulaciones fetales que descapsularon fácilmente y al corte mostraron una relación corticomedular normal. Los hallazgos más importantes fueron histopatológicos, traducidos en la presencia de numerosas vacuolas similares a las descritas en el hígado en cuanto a tamaño y forma, localizadas principalmente en los túbulos contorneados distales y proximales (Ver Figura 4). De manera similar al tejido hepático fueron positivas para coloraciones histoquímicas de grasa y negativas para glucógeno y moco.
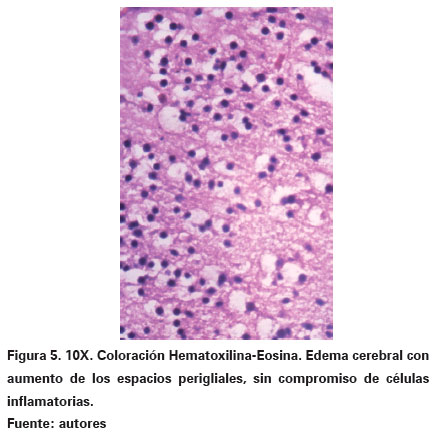
El cerebro, de 75 gramos, mostraba incremento del peso y volumen con presencia de circunvoluciones muy aplanadas y tumefactas, y surcos estrechos con cono de presión en amígdalas cerebelosas. Los cambios microscópicos mostraron aumento de los espacios perineuronales, perigliales y dilatación de los espacios perivasculares, lo que concuerda con el diagnóstico de edema cerebral (Ver Figura 5).
Los pulmones estaban aumentados de volumen, globosos y condensados con una imagen a la superficie de corte de hepatización roja con abundante salida de material de aspecto serosanguinolento. Histológicamente había consolidación hemorrágica, con vasos sanguíneos completamente congestivos y algunos bronquiolos mostraban eritrocitos intraluminales. Dentro de las luces alveolares se encontró abundante líquido de edema, eritrocitos y escasas células inflamatorias.
En la cavidad torácica no se observaron alteraciones congénitas de las vísceras. A nivel cardíaco no se encontraron alteraciones macroscópicas. El corazón, desde el punto de vista microscópico, mostraba congestión de los vasos sanguíneos con relativa extravasación de eritrocitos hacia el haz de His, que es el sustento morfológico de pacientes que presentan arritmias cardíacas. Dentro de las fibras miocárdicas no se evidenció infiltrado inflamatorio. Al corte de los diferentes órganos y tejidos se encontró al intestino, el páncreas, el bazo y el timo dentro de límites normales. En este caso se observaron abundantes linfocitos maduros pero no se visualizaron acúmulos linfoides.
DISCUSIÓN
Se describe el caso de una recién nacido de sexo masculino con buena adaptación neonatal, un examen físico y neurológico normal al nacer. Requirió a las 16 horas de su nacimiento la administración de gammaglobulina específica por antecedente de varicela materna, y terminó manifestándose como una muerte súbita sin respuesta al uso de corticoides, ni respuesta a la reanimación cardiopulmonar avanzada.
Para el diagnóstico del síndrome de Reye, se deben cumplir los tres criterios diagnósticos establecidos por el CDC27 que son: una encefalopatía aguda no inflamatoria documentada clínicamente por alteración del nivel de conciencia, y si es realizable, una biopsia cerebral con edema sin inflamación meníngea o perivascular o un análisis de líquido cefalorraquídeo con ocho o menos glóbulos blancos por milímetro cúbico; además debe tener hepatopatía con infiltración grasa hepática ya sea por biopsia o necropsia; y por último ninguna otra explicación para el edema cerebral o anormalidad hepática. En este caso se cumplen todos los criterios establecidos por el CDC debido a que se encontró en el examen histopatológico del hígado un compromiso vacuolar que correspondía a grasa en los citoplasmas de los hepatocitos, en el cerebro se evidenció edema cerebral a nivel macro y microscópicamente y no se encontró ninguna otra explicación razonable de las alteraciones neurológicas o hepáticas. En este recién nacido el síndrome de Reye estaba asociado a la infección por varicela adquirida de la madre, por transmisión vertical, caso no descrito previamente en la literatura. No se realizó punción lumbar porque el paciente no mostró clínica que indicara estudio del líquido cefalorraquídeo, como tampoco se llevó a cabo estudio de microscopia electrónica.
La fisiopatología más probable es la siguiente: un recién nacido cuya madre presentó varicela en las 37 5/7 semanas de gestación, quien sufrió viremia la cual desencadenó el edema cerebral y la lesión hepática y renal configurándose un diagnóstico de un síndrome de Reye congénito.
Los investigadores del estudio 'Reye's syndrome in the United States from 1981 through 1997' describieron a 1207 pacientes menores de 18 años con síndrome de Reye, donde encontraron que el 93,1% de los pacientes presentaron una infección viral hasta tres semanas antes del inicio del síndrome de Reye, el 73,4% tuvieron antecedente de una infección del tracto respiratorio superior y el 20,8% tuvieron antecedente de varicela8. No se han publicado casos de síndrome de Reye en recién nacidos asociado a varicela.
Se ha encontrado que el 13,5% de los pacientes con síndrome de Reye fueron menores de un año8. Peter Huttenlocher7 revisó el síndrome de reye en 59 menores de 10 meses con diagnóstico de síndrome de reye, donde encontró que el 53% eran de sexo femenino, el 50% de los casos ocurrieron entre el cuarto y quinto mes, el 86% presentaron vómito, el 98% convulsiones, el 91% dificultad respiratoria y el 61% fallecieron. Los exámenes de laboratorio evidenciaron aumento de dos veces del aspartato aminotransferasa en el 100%, la glucosa era menor de 50 mgr/dL en el 79% y el tiempo de protrombina fue menor de 75% en el 91% de los casos. Los hallazgos patológicos evidenciaron esteatosis hepática en el 100% de los niños.
Los infantes de mayor edad presentaban un pródromo viral leve y vómito, mientras que los lactantes estuvieron frecuentemente acompañados por diarrea. El coma, las convulsiones y las alteraciones de la respiración fueron las manifestaciones clínicas más frecuentes en lactantes. Las alteraciones de la respiración incluían hiperventilación o episodios a repetición de apnea. Las convulsiones fueron de gran mal, multifocales o mioclónicas. La hipertensión intracraneal manifestada por la fontanela anterior hipertensa fue en ocasiones más severa en niños mayores y terminaba en un edema cerebral fatal. Sin embargo, los lactantes fueron más susceptibles al daño directo derivado de las alteraciones metabólicas que ocurrieron en el síndrome de Reye. La mortalidad en lactantes fue mayor que en niños mayores.
Existe hasta la fecha un caso descrito de síndrome de Reye de etiología indeterminado en un recién nacido. Apostolos Papageorgiou28 en 1973 describió el caso de un neonato, hijo de madre de 28 años con 40 semanas de gestación. El embarazo fue reportado como no complicado, el parto fue vaginal espontáneo sin complicaciones con Apgar de 10 al minuto y de 10 a los cinco minutos. El recién nacido se encontró activo y normal hasta las 40 horas de edad cuando presentó taquipnea y tirajes. Se le diagnosticó una neumonía leve en bases pulmonares evidenciada en las radiografías y se trató con penicilina G. El recién nacido a las 48 horas de edad se encontró estuporoso, con opistótonos y fontanela abombada. La punción lumbar fue traumática y los hemocultivos fueron negativos. Los exámenes de laboratorios evidenciaron una hipoglicemia, hiperpotasemia, sodio sérico normal, cloro sérico normal y una acidosis metabólica. Se adicionó kanamicina al tratamiento y se inició infusión de dextrosa más bicarbonato. Después de 24 horas el paciente persiste con acidosis, presenta hepatomegalia y respiración de Cheyne Stokes con episodios de apneas. Se traslada a unidad de cuidados intensivos a las 78 horas de edad en donde se halló el paciente deshidratado, con dificultad respiratoria, pupilas dilatadas fijas, cianosis generalizada, arreflexia y pulsos no palpables. Presentó aminoacidemia, lactato sérico elevado, persistió con hipoglicemia, hiperpotasemia, acidosis metabólica y un cuadro hemático normal. Los cultivos del líquido cefalorraquídeo fueron normales. El paciente no responde al tratamiento instaurado y fallece a las 100 horas de vida. Se realiza una autopsia a las 25 horas después de la muerte en donde se encontró el hígado con tonalidad amarillenta con 99 gr de peso. Los riñones presentaron una débil tonalidad amarillenta con pesos normales. Al examen histopatológico se evidenció microvacuolas en las células hepáticas positivas para grasa en las tinciones Sudan Black B y Oil Red O. Los riñones presentaron los túbulos proximales finamente vacuolados con tinción Oil Red O positivas. El cerebro presentó un edema focal de la sustancia blanca y edema del citoplasma astrocítico. El miocardio evidenció áreas largas irregulares de gotitas finas de lípidos. Los cultivos bacteriológicos post mortem de la sangre aspirada de los pulmones, corazón y bazo fueron negativos. El autor concluye que este caso corresponde a un síndrome de Reye atribuido a algún evento intrauterino indeterminado tóxico, infeccioso o predisposición genética que fue responsable del curso postnatal inmediato. Se evidenció similitud en los hallazgos histopatológicos encontrados en el cerebro, hígado y riñón de este estudio con el actual caso.
Los diagnósticos diferenciales del síndrome de Reye incluyen meningitis, encefalitis, diabetes, sobredosis de medicamentos, síndrome de muerte súbita infantil, ingestión de tóxicos, trauma cráneo encefálico, envenenamiento, falla renal, falla hepática y desordenes congénitos del metabolismo8,29-31.
El paciente no mostró signos meníngeos ni signos de focalización neurológica, que junto a los hallazgos histopatológicos del cerebro permitió descartar una meningitis y encefalitis. Tampoco existió antecedente previo de ingesta alimentaria, envenenamiento, intoxicación y trauma craneal. Por la rápida instauración de sus síntomas y la muerte súbita, no se realizó exámenes de función hepática y renal.
En este caso la madre presentó varicela en el periodo comprendido entre cinco días antes y dos días después del parto, lo cual es una indicación de gammaglobulina específica con el fin de disminuir la posibilidad de varicela hemorrágica la cual tiene una mortalidad del 30%26. La varicela hemorrágica, es una forma grave de varicela donde las vesículas tienen contenido hemorrágico, hay petequias, sufusiones hemorrágicas, trombocitopenia y coagulación intravascular diseminada32.
Las reacciones adversas de la gammaglobulina específica son principalmente dolor, enrojecimiento y edema en el sitio de la aplicación, malestar general y erupciones. Las reacciones severas son poco comunes y pueden incluir dolor retroesternal, sensación constrictiva, disnea (uno de cada 500 casos), edema angioneurótico y shock anafiláctico (uno en 1 000 casos); esto es mencionado para la población general sin referirse específicamente a recién nacidos33.
Se habla de shock anafiláctico cuando este es mediado por la inmunoglobulina E, es decir, cuando el paciente es alérgico a determinado antígeno. Siempre tiene que haber una exposición previa para que el paciente se sensibilice y luego haya una reacción de respuesta a esa provocación que ha desarrollado anticuerpos específicos contra determinado antígeno. Aquí es de señalar que el recién nacido difícilmente ha tenido contacto previamente con los elementos que contienen una inmunoglobulina de uso endovenoso.
En los recién nacidos los niveles de inmunoglobulina E son prácticamente inexistentes, lo mismo ocurre con los niveles de complemento C3 y C5; por lo tanto el mecanismo inmunológico por el cual se desarrolla una anafilaxia no se presenta en un recién nacido. Un ejemplo de esto es que no se ha descrito una reacción anafiláctica en 5 000 aplicaciones en recién nacidos en estudios sobre la seguridad del uso de gammaglobulina específica para varicela34-35.
Las manifestaciones clínicas de la anafilaxia comprenden urticaria, angioedema de párpados, labios o epiglotis, conjuntivitis, lagrimeo, rinorrea, estornudos, broncoespasmo, retortijón abdominal, náuseas, vómito, diarrea, arritmias e hipotensión. A pesar de que en este caso el paciente presenta hipotensión, no se relatan manifestaciones en piel, angioedema u obstrucción de la vía aérea, por lo que no existe manera de hacer diagnóstico clínico de anafilaxia36,8.
Otra manera de enfocar este caso es desde el punto de vista de causas de muerte súbita. El síndrome de muerte súbita en un recién nacido se ha asociado a algunos factores de riesgo como la posición en prono, entre otros. En este síndrome los recién nacidos son aparentemente sanos y el análisis cuidadoso de la escena de la muerte es negativo, es decir, siempre en estos casos hay que investigar el ambiente donde el niño se encontraba ya que donde fallece es donde se puede encontrar la explicación a la muerte39.
En la literatura se encuentra que la primera causa de muerte súbita en el recién nacido son los problemas cardiacos. En este paciente se descartan causas cardíacas de muerte súbita como la tetralogía de Fallot, la transposición de grandes vasos, la estenosis aórtica, el síndrome de Marfán, el síndrome de Eisenmenger y entre las adquiridas están la cardiomiopatía dilatada o la miocarditis39.
Como segunda causa de muerte súbita en el recién nacido se ubican las infecciones, siendo la mayoría virales o sepsis que habían pasado inadvertidas. Se menciona que ninguno de estos pacientes presenta sintomatología previa, y que la manifestación inicial fue la muerte súbita; descripción que concuerda con este caso39.
Otra causa de muerte súbita son los errores congénitos del metabolismo, que generalmente conducen a situaciones de acidosis metabólica o de hipoglicemia. No se realizaron estudios para descartar estas patologías.
CONCLUSIÓN
El síndrome de Reye es una encefalopatía aguda y degeneración grasa del hígado, observada usualmente cinco a siete días después una infección del tracto respiratorio o varicela. Se describe el caso de un recién nacido con antecedente de varicela materna que presentó un síndrome de Reye congénito y terminó manifestándose clínicamente como muerte súbita; aspecto que es muy llamativo por el inicio abrupto, rápido y severo de los síntomas y signos. Cabe resaltar que no se han publicado casos de síndrome de Reye congénito asociado a varicela, y que podría ser la primera descripción hasta la fecha.
CONFLICTOS DE INTERÉS
Los autores manifiestan que no hubo conflictos de interés, ni tampoco hubo fuente de financiación externa.
REFERENCIAS BIBLIOGRÁFICAS
1. Reye RDK, Morgan G, Baral J. Encephalopathy and fatty degeneration of the viscera: a disease entity in childhood. Lancet. 1963;2:749. [ Links ]
2. Balistreri WF. Reye syndrome and "reye - like" diseases. In: Behrman RE, Kliegman RM, Arvin AM, editors. Nelson Textbook of Pediatrics. 15th ed. Philadelphia: W.B. Saunders Company; 1996. p 1144-5. [ Links ]
3. McGovern M, Glasgow J, Stewart M. Lesson of the week: Reye's syndrome and aspirin: lest we forget. BMJ. 2001;322:1591-2. [ Links ]
4. Hurwitz ES, Nelson DB, Davis C, Morens D, Schonberger LB. National surveillance for Reye syndrome: A five year review. Pediatrics. 1982;70:895. [ Links ]
5. Trauner DA. Reye's syndrome [Medical Progress]. West J Med. 1984;141:206-9. [ Links ]
6. Lovejoy FH Jr, Smith AL, Bresnan MJ, Wood JN, Victor DI, Adams PC. Clinical Staging in Reye Syndrome. Am J Dis Child. 1974;128(1):36-41. [ Links ]
7. Huttenlocher PR, Trauner DA. Reye's syndrome in infancy. Pediatrics. 1978;62(1):84-90. [ Links ]
8. Belay ED, Bresee JS, Holman RC, Khan AS, Shahriari A, Schonberger LB. Reye's syndrome in the United States from 1981 through 1997. N Engl J Med. 1999;340(18):1377-82. [ Links ]
9. Baldellou A. Síndrome de Reye. Cuarenta años después. An Pediatr (Barc). 2003;59(4):319-22. [ Links ]
10. Daugherty CC. A morphometric study of Reye's Syndrome. Am J Path. 1987;129:313-6. [ Links ]
11. Weiner DL. Reye síndrome: http://emedicine.medscape.com/article/803683-overview [ Links ]
12. Benítez L, Pretelt F, Morillo L. Síndrome de Reye: Informe de un caso en adultos. Acta Medica Colombiana. 1992;17(1):63-7. [ Links ].
13. Casteels-Van Daele M, Van Geet C, Wouters C, Eggermont E. Reye syndrome revisited: a descriptive term covering a group of heterogeneous disorders. Eur J Pediatr. 2000;159(9):641-8. [ Links ]
14. Castillo F. Ácido acetilsalicílico y síndrome de Reye. Offarm 2003;22(9):167. [ Links ]
15. Prandota J. Important role of prodromal viral infections responsible for inhibition of xenobiotic metabolizing enzymes in the pathomechanism of idiopathic Reye's syndrome, Stevens- Johnson syndrome, autoimmune hepatitis, and hepatotoxicity of the therapeutic doses of acetaminophen used in genetically predisposed persons. Am J Ther. 2002;9(2):149-56. [ Links ]
16. Schrör K. Aspirin and Reye Syndrome A Review of the Evidence. Pediatr Drugs. 2007;9(3):195-204. [ Links ]
17. Reye syndrome-insights on causation and prognosis. Arch Dis Child. 2001;85:351-3. [ Links ]
18. Mowat AP. Reye's syndrome and aspirin. In: Vane JR, Botting RM, editors. Aspirin and other salicylates. London: Chapman & Hall Medical. 1992:531-47. [ Links ]
19. Corey L, Rubin RJ, Hattwick MAW, Noble GR, Cassidy E. A nationwide outbreak of Reye's syndrome: its epidemiologic relationship to influenza B. Am J Med. 1976;61:615-25. [ Links ]
20. Starko KM, Ray CG, Dominguez LB, Stromberg WL, Woodall DF. Reye's syndrome and salicylate use. Pediatrics. 1980;66:859-64. [ Links ]
21. Waldman RJ, Hall WN, McGee H, Van Amburg G. Aspirin as a risk factor in Reye's syndrome. JAMA . 1982;247:3089-94. [ Links ]
22. Hurwitz ES, Barrett MJ, Bregman D, Gunn WJ, Schonberger LB, Fairweather WR, et al. Public Health Service study of Reye's syndrome and medications: report of the main study. JAMA. 1987;257:1905-11. [ Links ]
23. Duerksen D, Jewell L, Mason A, Bain V. Co-existence of hepatitis A and adult Reye's. Gut. 1997;41:121-4. [ Links ]
24. Stumpf DA. Reye syndrome: an international perspectiva. Brain & Development. 1995;17(suppI):77-8. [ Links ]
25. Felipo V, Butterworth R. Mitochondrial dysfunction in acute hyperammonemia. Neurochemistry International. 2002;40:487-91. [ Links ]
26. American Academy of Pediatrics. Varicella-zoster Infections. In: Pickering LK, Baker CJ, Long SS, Kimberlin DW, editors. Red Book: 2012 Report of the Committee on Infectious Diseases. 29th ed. Elk Grove Village: American Academy of Pediatrics; 2012. p.774-89. [ Links ]
27. CDC. Reye Syndrome 1990 Case Definition: http://wwwn.cdc.gov/nndss/script/casedef.aspx?condyrid=821&datepub=1/1/1990%2012:00:00%20am . [ Links ]
28. Papageorgiou A, Wiglesworth FW, Schiff D, Stern L. Reye's syndrome in a newborn infant. Can Med Assoc J. 1973;109(8):717-20. [ Links ]
29. National Reye's Syndrome Foundation. What is Reye's Syndrome?: http://www.reyessyndrome.org/what.html [ Links ]
30. Quintillá J, Campistol J, Boleda M, Vilaseca M, Artuch R, Palomeque A, et al. Síndrome de Reye-like como manifestación inicial de enfermedad mitocondrial. An Esp Pediatr. 2000;52:479-82. [ Links ]
31. Burton B. Inborn Errors of Metabolism in Infancy: A Guide to Diagnosis. Pediatrics. 1998;102:69. [ Links ]
32. Corrigan J, Watkins L. Hemorrhagic chickenpox associated with disseminated intravascular coagulation. Pediatric Research 1971;5:404-5. [ Links ]
33. American Academy of Pediatrics. Passive immunization. In: Pickering LK, Baker CJ, Long SS, McMillan JA, editors. Red Book: 2012 Report of the Committee on Infectious Diseases. 29th ed. Elk Grove Village: American Academy of Pediatrics; 2012. p.56-69. [ Links ]
34. Ohlsson A, Lacy J. Intravenous immunoglobulin for suspected or subsequently proven infection in neonates. Cochrane Database Syst Rev. 2010;17(3):CD001239. [ Links ]
35. Ohlsson A, Lacy JB. Intravenous immunoglobulin for preventing infection in preterm and/or low-birth-weight infants. Cochrane Database Syst Rev. 2004;(1):CD000361. [ Links ]
36. Cardona R, Montoya F, Orrego JC. Anafilaxia. Iatreia. 2000;13(1): 16-31. [ Links ]
37. Shearer W, Fleisher T. Constituyent and Development of the Inmune System. In: Middleton E, editors. Alergy: principIes and practice. 5th ed. St Louis: Mosby; 1998. p.2-8. [ Links ]
38. Gold Y, Golderg A, Sivan Y. Hyper-releasability of mast cells in family members of infants with sudden infant death syndrome and apparent life-threatening events. J Pediatr. 2000;136(4):460-5. [ Links ]
39. Weber MA, Ashworth MT, Risdon RA, Brooke I, Malone M, Sebire NJ. Sudden unexpected neonatal death in the first week of life: autopsy findings from a specialist centre. J Matern Fetal Neonatal Med. 2009;22(5):398-404. [ Links ]
