










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.27 no.3 Bicaramanga Sep./Dec. 2014
Miocardiopatía arritmogénica del ventrículo
derecho
Alexander Álvarez-Ortiz1
Sergio Navas Gutiérrez1
Erika Martínez2
Marco De León3
Leonor Mariño-Murillo4
Victor Manuel Mora-Bautista4
Héctor Hernández Gallo5
Hedilberto Duarte Hernández6
Manuel Guillermo Hernández6
Jaime Gómez Ayala7
Sergio Andrés Higuera Leal8
1Médico. Internista Cardiólogo Electrofisiólogo. Instituto del Corazón de Bucaramanga. Bucaramanga. Santander. Colombia.
2Médico. Internista Residente de segundo año de Cardiología. Universidad El Bosque. Bogotá. Colombia.
3Médico. Internista Residente de segundo año de Cardiología. Universidad El Rosario. Bogotá. Colombia.
4Médico. Clínica Chicamocha. Bucaramanga. Santander. Colombia.
5Médico. Cardiólogo Hemodinamista. Instituto del Corazón de Bucaramanga. Bucaramanga. Santander. Colombia
6Médico. Internista Cardiólogo Ecocardiografista. Instituto del Corazón de Bucaramanga. Bucaramanga. Santander. Colombia.
7Médico. Internista. Instituto del Corazón de Bucaramanga. Bucaramanga. Santander. Colombia.
8Médico. S.S.O. Instituto del Corazón de Bucaramanga. Bucaramanga. Santander. Colombia.
Correspondencia: Dr. Alexander Álvarez Ortiz. Instituto del Corazón de Bucaramanga, carrera 28 N° 40-05 Mejoras Públicas Bucaramanga. Santander.
Colombia. Correo electrónico: alalort@hotmail.com
Artículo recibido 12 enero de 2014 y aceptado para publicación 30 de junio de 2014
RESUMEN
La miocardiopatía arritmogénica del ventrículo derecho es una patología, en la mayoría de los casos de origen genético autosómico dominante caracterizado por el compromiso, tanto morfológico como funcional, del ventrículo derecho en el que se reemplaza el tejido del miocardio normal por tejido fibroadiposo, generando un sustrato arritmogénico. Se debe sospechar en todo paciente joven que presente síncope, taquiarritmia ventricular o paro cardiaco. Su diagnóstico se establece por la sumatoria de criterios que incluyen hallazgos morfológicos, electrocardiográficos y alteraciones funcionales. En la actualidad no hay un tratamiento único establecido; sin embargo, se sigue trabajando en el diagnóstico temprano y el uso de terapias más avanzadas. Se realiza una revisión de la literatura en el contexto de la presentación de un caso clínico diagnosticado en la ciudad de Bucaramanga en un adulto joven de género masculino. MÉD.UIS. 27(3):123-134.
Palabras Clave: Displasia Ventricular Derecha Arritmogénica. Taquicardia ventricular. Fibrilación ventricular. Síncope.
Arrytmogenic right ventricular dysplasia
ABSTRACT
Arrhythmogenic right ventricular dysplasia is a pathology, mostly genetic of dominant autosomic pattern characterized by both morphologic and functional compromise of the right ventricle in which normal myocardial tissue its replaced by fibrous and adipose tissue generating an arrhythmogenic substrate. It must be evaluated in all young patients presenting syncope, ventricular tachyarrhythmia or cardiac arrest. Its diagnosis it's established upon the consideration of morphological criteria, electrocardiographic findings and functional alterations. Currently there is not a definite treatment established; however there is ongoing research in early diagnosis and advanced therapies usage. In this article we provide a literature review in the context of a clinical case diagnosed in a male young adult from the city of Bucaramanga in Colombia. MÉD.UIS. 27(3):123-134.
Keywords: Arrhythmogenic right ventricular dysplasia. Ventricular tachycardia. Ventricular fibrillation. Syncope.
¿Como citar este artículo?: Álvarez-Ortiz A, Navas S, Martínez E, De León M, Mariño-Murillo
L, Mora-Bautista VM, et al. Miocardiopatía arritmogénica del ventrículo derecho. MÉD.UIS.
27(3):123-134.
INTRODUCCIÓN
La displasia arritmogénica del ventrículo derecho es una forma de miocardiopatía en la cual se generan cambios en la composición celular del ventrículo derecho dandose un reemplazo de los miocitos normales por tejido fibroadiposo y que finalmente lleva a la generación de un sustrato arritmogénico, pudiendo inclusive llevar a síncope y arritmias malignas. Es una enfermedad que puede presentarse hasta en el 0,8% de la población dependiendo de las áreas geográficas. En este artículo se presenta un caso de displasia arritmogénica del ventrículo derecho atendido en un centro de cardiología de alta complejidad en la ciudad de Bucaramanga y se hace una revisión de los aspectos fisiopatológicos, diagnósticos y terapéuticos actuales con el objetivo de mejorar el conocimiento de la patología y de esta manera impactar en su atención por parte del personal clínico.
PRESENTACIÓN DEL CASO CLÍNICO
Adulto joven de 36 años con múltiples consultas al servicio de urgencias por episodios de dolor torácico, disnea, deterioro de la clase funcional (clasificación funcional New York Heart Association II - III), astenia, presíncope recurrente, adinamia y sensación de palpitaciones frecuentes; los cuales iniciaron dos meses antes de la presentación. Sin antecedentes cardiovasculares personales ni familiares, sólo refirió una laparotomía de emergencia por ruptura hepática luego de trauma cerrado de abdomen. A la evaluación de ingreso se encontró sin alteración del estado de conciencia, con cifras tensionales dentro de límites normales, con frecuencia cardiaca y respiratoria normales. A la auscultación cardiopulmonar sin soplos cardíacos pero se documentó un reforzamiento del segundo ruido en foco pulmonar. El resto del examen físico fue normal.
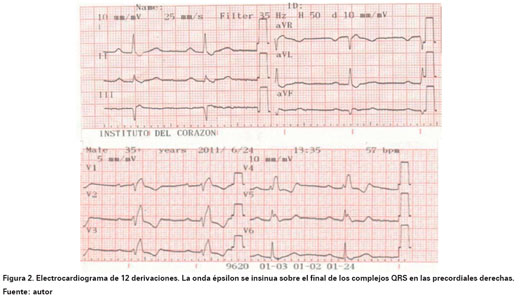
Por las características clínicas y su recurrencia, fue hospitalizado para estudios complementarios. Inicialmente se realizó una radiografía de tórax evidenciando cardiomegalia global de predominio derecho, cefalización del flujo pulmonar, signos de hipertensión pulmonar pre y poscapilar, sin otros hallazgos en los campos pulmonares (Ver Figura 1). El electrocardiograma de superficie de 12 derivaciones mostró ritmo sinusal, bloqueo aurículoventricular de primer grado, bloqueo de rama derecha completo, patrón de necrosis en la cara inferior e imagen sugestiva de onda épsilon (Ver Figura 2). El ecocardiograma transtorácico bidimensional evidenció aquinesia apical del ventrículo derecho sugestiva de un infarto ventricular derecho, insuficiencia tricuspídea severa, dilatación severa de la aurícula derecha, y ventrículo izquierdo con estructura y función sistólica y diastólica normal (Ver Figura 3).


Por los hallazgos ecocardiográficos se consideraron tres posibilidades diagnósticas: tromboembolismo pulmonar submasivo, enfermedad coronaria y Miocardiopatía Arritmogénica del Ventrículo Derecho (MAVD). Se iniciaron estudios complementarios para determinar la etiología de su cardiopatía. Se realizó tomografía de tórax con protocolo para tromboembolismo pulmonar sin evidencia de defectos de llenado a nivel vascular pulmonar, descartándose este diagnóstico.
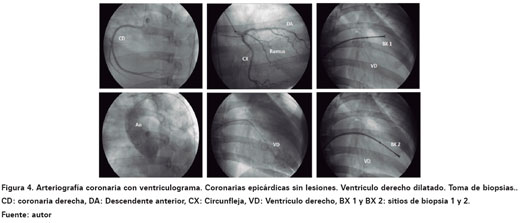
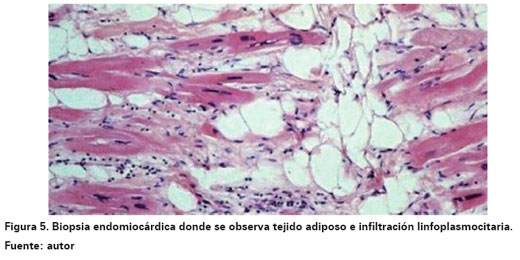
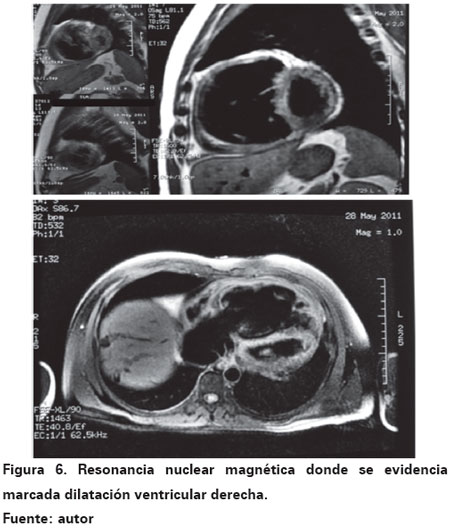
La arteriografía coronaria con biopsia endomiocárdica evidenció arterias coronarias epicárdicas sin lesiones angiográficamente significativas, lo que excluyó la enfermedad coronaria. El ventriculograma derecho mostró dilatación severa de cavidades derechas con insuficiencia tricuspídea, dilatación de tracto de salida y del tronco pulmonar sin otras alteraciones angiográficas. Se envió la muestra endomiocárdica para estudio histopatológico (Ver Figura 4). La biopsia endomiocárdica fue descrita con presencia de fibras miocárdicas con cambios de hipertrofia, agrandamiento nuclear e hipercromatismo, focos de hipertrofia miocítica y escasa infiltración fibroadiposa, con leve infiltración neutrofílica y linfoplasmocitaria (Ver Figura 5). Posteriormente, se realizó una resonancia magnética torácica y cardíaca mostrando miocardiopatía dilatada que afecta cavidades derechas con insuficiencia severa de la válvula tricúspide (Ver Figura 6). Debido a que el paciente refería episodios de taquicardia, se realizó un holter electrocardiográfico de 24 horas documentándose 3589 extrasístoles ventriculares monomórficas y múltiples episodios de taquicardia ventricular monomórfica no sostenida (Ver Figura 7). Por el cuadro clínico descrito y los hallazgos electrocardiográficos, imagenológicos y angiográficos se consideró que el paciente tenía una MAVD con severo compromiso de su función ventricular y alto riesgo de muerte súbita, por lo cual en junta médica se decidió el implante de un cardiodesfibrilador bicameral como prevención primaria e ingreso a clínica de falla cardiaca para continuar el seguimiento y titulación de terapia médica estándar para falla cardiaca.

DISCUSIÓN
El primer caso de MAVD fue descrito por Osler en 1905, quien reportó el caso de un hombre de 40 años que presentó muerte súbita cuando subía una colina, y en la necropsia se encontró un adelgazamiento de la pared del ventrículo derecho que fue descrita como "apergaminada", descartándose también patología coronaria y valvular1. Posteriormente en los años 50 se describen varios casos en menores de 20 años con insuficiencia cardiaca en quienes se evidenció adelgazamiento de la pared del ventrículo derecho. Sin embargo, solo hasta 1960 Dalla Volta hace una descripción de la patología y la llama auricularización del ventrículo derecho. Posteriormente Fontaine le da el nombre de "Displasia arritmogénica ventricular del ventrículo derecho" al describir seis casos de pacientes con taquicardia ventricular sostenida sin insuficiencia cardiaca, en un capítulo de libro2. Desde 1994 el diagnóstico clínico se basa en los criterios establecidos por la Sociedad Europea de Cardiología, la Sociedad Internacional y la Federación de Cardiología, publicados en el British Heart Journal. La MAVD es una enfermedad del miocardio genéticamente determinada, de patrón de herencia autosómica dominante, que se caracteriza por la presencia de arritmias y una serie de anomalías funcionales y estructurales del ventrículo derecho, debido a la sustitución progresiva de miocardio por tejido graso y fibroso, llevando a desenlaces fatales como la muerte súbita3. Es una patología con una tipificación relativamente joven, que tan solo hasta mediados de los 90 fue incluida en el subgrupo de miocardiopatías, por lo que hay un subregistro de la misma y su incidencia real se desconoce. La prevalencia de la enfermedad se estima entre 0,02 a 0,1% en la población general; sin embargo, depende de la ubicación geográfica: en ciertas regiones de Italia y Grecia se ha encontrado una prevalencia de 0,4 a 0,8%. Se presenta principalmente en adultos jóvenes, hasta un 80% en menores de 40 años, con una relación de hombre:mujer de 2,7:14. La mortalidad asociada a la patología oscila entre un 4 a 20%. De hecho, hasta el 5% de las muertes súbitas en adultos jóvenes en los EE.UU. y hasta el 25% de las muertes relacionadas con el ejercicio en la región de Véneto en Italia se atribuyen a la MAVD5.
La patogénesis de la MAVD no es completamente conocida. Sin embargo, la agregación familiar representa hasta el 50% de los casos, con un patrón herencia en su mayoria autosómica dominante con expresión variable y penetrancia incompleta6. Hasta la fecha se han identificado ocho genes relacionados directamente con la enfermedad, incluyendo el gen DSP que codifica para desmoplakina, el PKP2 que produce la plakofilina 2, el DSG2 de la desmogleína 2, el DSC2 correspondiente a la desmocolina, el JUP de la plakoglobina, el TGTB3 o Factor de crecimiento transformante B3, el TMEM43 que es transcrito para formar proteína transmembrana 43 y el TP63 que secuencia la proteína tumoral 637. Cinco de estos genes (DSP, PKP2, DSC2, DSG2, JUP) codifican para proteínas desmosómicas, cuyas funciones son la adhesión celular, adipogénesis, apoptosis, regulación de uniones y homeostasis del calcio.
Las proteínas más estudiadas en la MAVD son la plakoglobina y la desmoplaquina. La primera es una proteína citoplasmática que participa en la unión de los filamentos intermedios y el citoesqueleto de la actina con los complejos transmembrana que conectan los complejos adyacentes. Por otra parte la desmoplaquina ancla los filamentos intermedios a la membrana plasmática y constituye una base para el mantenimiento de la integridad celular, por lo que un error en la codificación de alguna de ellas llevara a un sustrato inestable8. Se han puntualizado seis mutaciones en el receptor cardiaco de la rianodina 2, las que generarían un aumento en la concentración citosólica de calcio, siendo proapoptóticas.
Adicional al componente genético descrito, se ha encontrado en estos pacientes una respuesta catecolaminérgica aumentada que genera taquicardias ventriculares polimórficas, las cuales se han descrito como formas primarias de MAVD7. Sin embargo, las mutaciones demuestran penetrancia incompleta y expresividad variable, lo que sugiere que factores ambientales también están implicados en la patogénesis8.
Entre otras teorías patogénicas se encuentran: la teoría inflamatoria, dado que en las biopsias se evidencian infiltrados celulares, por lo que los ciclos de daño y reparación continua generarían tejido fibroso que remplazaría el tejido miocárdico9. La teoría apoptótica sugiere que la sustitución progresiva del miocardio por tejido adiposo y fibroso sucedería luego de una apoptosis aumentada10. Adicionalmente, dentro de esta baraja de posibilidades se ha descrito el hallazgo de virus cardiotropos como el virus Coxsackie B3, algunos enterovirus y adenovirus11. A pesar de lo intrincadas que parecen, ninguna explica el desencadenante exacto de la MAVD.
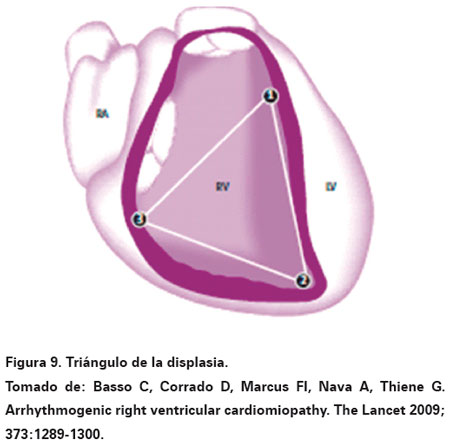
A nivel morfológico se evidencia dilatación del ventrículo derecho con pérdida difusa o segmentaria de los miocitos, los cuales son sustituidos por tejido fibroadiposo, evidenciándose además adelgazamiento de la pared y atrofia de los miocitos restantes (Ver Figura 8)12. El reemplazo fibroadiposo comienza por el epicardio y avanza hasta el endocardio, aunque el compromiso usualmente es del ventrículo derecho también puede comprometer al tabique interventricular y la pared libre del ventrículo izquierdo. Existe una zona de predilección para el reemplazo del tejido llamada el triángulo de la MAVD, formada por el tracto de salida del ventrículo derecho, el ápex y el infundíbulo (Ver Figura 9). En el área afectada se puede formar un agujero electrofisiológico que puede potencialmente constituirse en un sustrato para las arritmias de reentrada9. Para hacer el diagnóstico definitivo, la cantidad miocitos residuales en el tejido debe ser menor a 45%, si es de 45 a 70% es una forma limítrofe de MAVD, pero si es mayor a 70% este diagnóstico puede ser excluido13.

La presentación clínica es variable, depende de la inestabilidad cardÍaca y la disfunción ventricular, pudiendo expresar una gran variedad de síntomas como dolor torácico atípico, disnea, síncope, síndrome coronario agudo, signos de falla ventricular derecha o biventricular, taquicardias, taquicardias ventriculares sostenidas y muerte súbita. Para fines prácticos, todo este espectro de hallazgos se organizan en las siguientes cuatro fases:
- Fase temprana o silente: generalmente asintomática, aunque puede debutar con muerte súbita.
- Fase inestable: con predominio de arritmias sintomáticas. En el electrocardiograma puede presentar taquicardia y evidencia de morfología de bloqueo de rama izquierda, sugestiva de origen ventricular derecho.
- Fase de fallo ventricular derecho: con relativa conservación de la función ventricular izquierda
- Fase final: con progresiva dilatación biventricular14
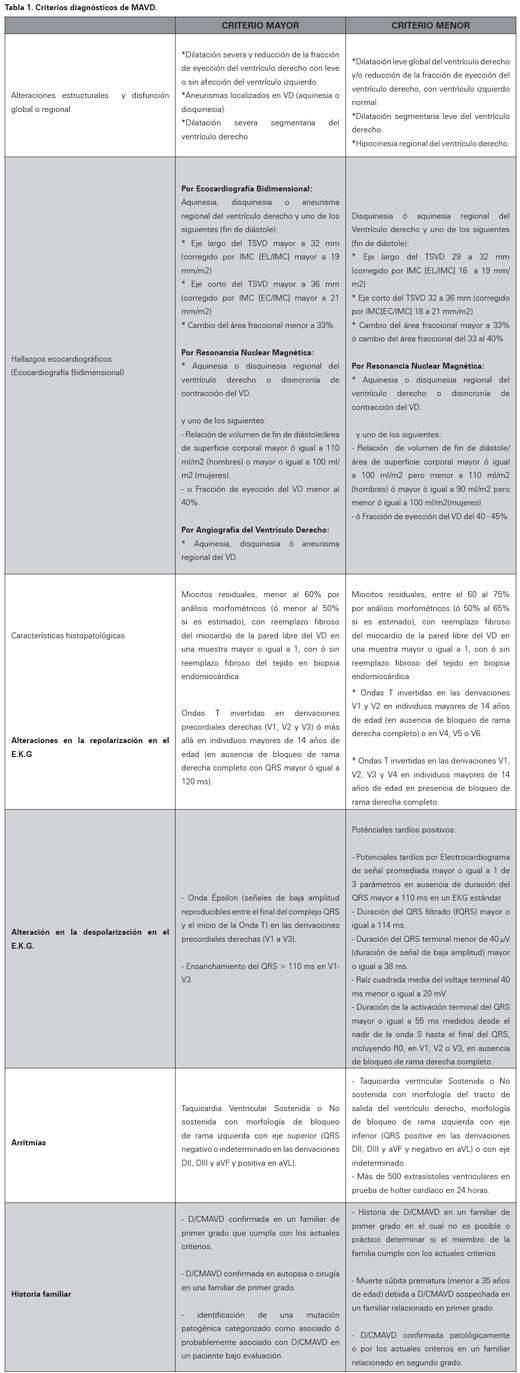
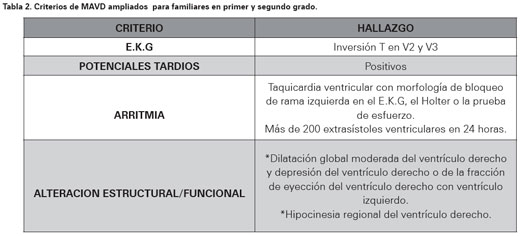
No existe una única prueba para establecer el diagnóstico de MAVD, por lo que se requiere una evaluación detallada clínica, funcional, morfológica y electrocardiográfica. Teniendo en cuenta lo anterior, en 1994 McKenna y colaboradores propusieron una serie de criterios mayores y menores (Ver Tabla 1)15. Para hacer el diagnóstico se deben cumplir dos criterios mayores, un criterio mayor y dos menores ó cuatro menores. Más adelante en el 2002, Hamid propone una serie de criterios para aumentar la sensibilidad diagnóstica en los familiares de primer grado (Ver Tabla 2)16.
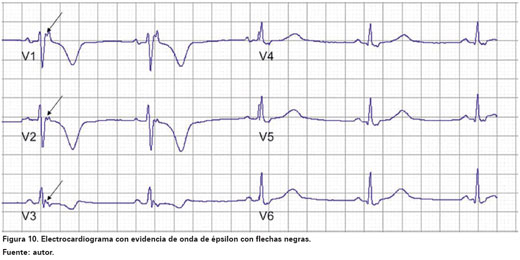
Las alteraciones electrocardiográficas se evidencian en un 90% de los pacientes con MAVD. Entre las más frecuentes se encuentran: inversión de la onda T, presente en 50% de los pacientes; bloqueo de rama derecha incompleto en 18% de los pacientes y de forma completa en 15%, además de prolongación del QRS mayor a 110 mseg en las derivaciones V1 y V2. Un hallazgo más específico de esta enfermedad es la presencia de una desviación de la mitad, de final del complejo QRS o inicio del segmento ST llamada como onda épsilon, presente en 30% de los pacientes (Ver Figura 10).
La ecocardiografía es la primera técnica no invasiva a utilizarse para la evaluación morfológica. Permite analizar las anomalías estructurales del ventrículo derecho encontrándose dilatación en la zona del llamado triángulo de la displasia, aneurismas telediastólicos, hipocinesia regional y adelgazamiento de la pared del ventrículo. Otros hallazgos menos frecuentes esta el aumento de la ecogenicidad de la banda moderadora, trabéculas prominentes en el ápex y prolapso de la válvula tricúspide17. La ecocardiografía permite no sólo evaluar la morfología, sino también llevar un registro de la progresión de la enfermedad, hacer diagnóstico temprano en los familiares y establecer diagnósticos diferenciales con otras patologías.
La resonancia magnética se ha convertido en la prueba de oro para el diagnóstico de la MAVD, dado que permite la visualización no solo de la morfología del ventrículo derecho sino también su funcionalidad y el flujo en tiempos dinámicos. Morfológicamente permite evaluar los depósitos de grasa intramiocárdica, adelgazamientos focales de la pared, desorden trabecular y la ampliación del tracto de salida del ventrículo derecho. Funcionalmente permite evaluar la presencia de aneurismas del ventrículo derecho, falla sistólica, la velocidad de flujo de la válvula tricuspídea, el cual puede ser un signo temprano de MAVD18. Esta técnica presenta como desventaja el hecho de ser observador dependiente19.
La ventriculografía derecha se considera la prueba de referencia para la evaluación de la función del ventrículo derecho, en ella se evidencia discinesia en el triángulo anatómico y la presencia de trabéculas hipertróficas20. Sin embargo, dada su naturaleza invasiva y su variación interobservador, esta prueba no está ampliamente difundida.
La biopsia endomiocárdica es uno de los criterios definitivos con la demostración histológica de la presencia de tejido fibroadiposo. Sin embargo, su utilidad es controversial dado el compromiso segmentario de la MAVD y el alto riesgo de perforación en la pared del ventrículo, lo que dificulta obtener una muestra adecuada.
Las manifestaciones arrítmicas de la enfermedad son variables. No se conoce el papel pronóstico del estudio electrofisiológico en los pacientes con complejos ventriculares prematuros aislados o taquicardia ventricular no sostenida. Di Biase y colaboradores21 evaluaron el estudio electrofisiológico en un número limitado de pacientes a fin de estratificar el riesgo. En 17 pacientes con displasia leve, indujo taquicardia ventricular solo en pacientes con taquicardia ventricular espontánea. En otro estudio la taquicardia ventricular fue inducida en 90% de 12 pacientes con taquicardia ventricular espontánea sostenida. El valor predictivo positivo para taquicardia ventricular recurrente fue sólo de 55%. La taquicardia ventricular sostenida no pudo ser inducida en 20 pacientes con taquicardia ventricular no sostenida. En este estudio la inducibilidad fue de 88% en pacientes con taquicardia ventricular sostenida. En general, el estudio electrofisiológico se utiliza para reproducir la taquicardia ventricular clínica y guiar la ablación. En conclusión, el estudio electrofisiológico podría ser útil en la evaluación del riesgo de muerte súbita cardiaca en pacientes con miocardiopatía arritmogénica del ventrículo derecho (recomendación IIb - nivel de evidencia C)22.
El tratamiento debe ser individualizado, entre las diferentes opciones de manejo se encuentra el tratamiento antiarrítmico y para falla cardíaca, ablación con energía de radiofrecuencia, terapia con cardiodesfibrilador implantable y el tratamiento quirúrgico.
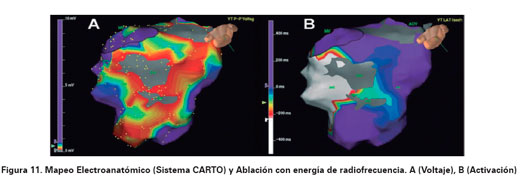
El tratamiento farmacológico está orientado al manejo y prevención de las arritmias y de la falla cardiaca. Para el manejo de las arritmias se pueden utilizar betabloqueadores, amiodarona y sotalol, este último usado en dosis elevadas ha mostrado superioridad frente a los dos primeros, pero en dosis bajas no existen diferencias entre los tres23. La amiodarona o el sotalol pueden ser efectivos para el tratamiento de la taquicardia ventricular sostenida o la fibrilación ventricular en pacientes con MAVD cuando no sea posible el implante de un cardiodesfibrilador (recomendación IIa- nivel de evidencia C). Cuando la enfermedad ha progresado a falla cardiaca derecha o biventricular el manejo farmacológico consiste en betabloqueadores, IECA, diuréticos, antagonista de la aldosterona y anticoagulantes9. La ablación con energía de radiofrecuencia puede ser útil como terapia adyuvante en el manejo de los pacientes que tengan taquicardia ventricular recurrente a pesar de terapia antiarrítmica óptima (recomendación IIa - nivel de evidencia C, Ver Figura 11).
Se recomienda el implante de cardiodesfibrilador para la prevención de muerte súbita cardiaca en pacientes con MAVD con taquicardia ventricular sostenida o fibrilación ventricular documentadas (recomendación Clase I - nivel de evidencia B). Puede ser efectivo para la prevención de muerte súbita cardiaca en pacientes que tengan enfermedad extensa, incluyendo aquellos con compromiso del ventrículo izquierdo, uno o más familiares afectados con muerte súbita cardiaca o síncope inexplicado, cuando se han excluido la taquicardia ventricular o la fibrilación ventricular como la causa del síncope (recomendación IIa - nivel de evidencia C). Ambos grupos de personas deben recibir terapia médica óptima de manera crónica y tener una expectativa de sobrevida razonable con un buen estado funcional por más de un año.
Hace más de 20 años, el tratamiento quirúrgico fue introducido inicialmente con el objetivo de realizar ventriculotomía derecha. La desconexión total de la pared libre derecha también fue probada, no obstante, ésta aproximación fue abandonada por múltiples razones: riesgo de desarrollar insuficiencia cardíaca derecha, incremento en la disponibilidad de trasplante cardíaco y el implante de cardiodesfibrilador. Ocasionalmente, la cirugía de desconexión ó la cardiomioplastia del ventrículo derecho se usan como alternativas terapéuticas.
CONCLUSIÓN
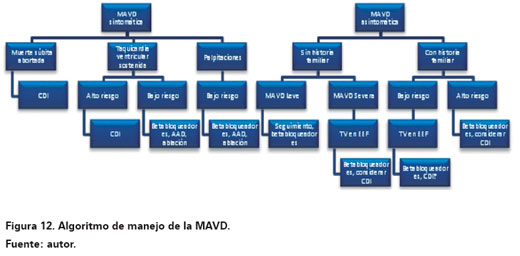
En el caso clínico presentado se optó por hacer el diagnóstico diferencial con enfermedad coronaria y tromboembolismo pulmonar por el compromiso de novo de cardiomiopatía dilatada con compromiso severo del ventrículo derecho. Aunque la biopsia endomiocárdica no es absolutamente mandatoria para hacer el diagnóstico, fue realizada por la indicación de la arteriografía coronaria, aprovechando el acceso vascular durante este examen. Solo se colocó el cardiodesfibrilador sin pensar en la opción quirúrgica por la estabilización clínica con el tratamiento médico, dado que esta opción es un tratamiento de tercera línea. Se presenta el algoritmo aceptado de manejo para esta patología, que condensa las recomendaciones mencionadas y bajo el cual fue manejado el paciente del caso clínico (Ver Figura 12).
REFERENCIAS BIBLIOGRÁFICAS
1. Osler W. The Principles and Practice of Medicine. 6th Ed. New York: Appleton Century Crofts, 1905: 820. [ Links ]
2. Marcus FI, Fontaine G, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982; 65:384- 99. [ Links ]
3. Gear K, Marcus FI. Arrhythmogenic right ventricular dysplasia / cardiomyopathy. Circulation. 2003; 107: e31-3. [ Links ]
4. Thiene G, Basso C. Arrhythmogenic right ventricular cardiomyopathy: an update. Cardiovasc Pathol. 2001; 10:109 -17. [ Links ]
5. Kies P, Bootsma M, Bax J, Schalij MJ, van der Wall EE. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: screening, diagnosis, and treatment. Heart Rhythm. 2006; 3(2): 225- 34. [ Links ]
6. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom- Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia / cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994; 71:215-8. [ Links ]
7. van der Zwaag P, Jongbloed J, van den Berg M, van der Smagt J, Jongbloed R, Bikker H, et al. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum. Mutat. 2009; 30 (9): 1278-83. [ Links ]
8. Awad M, Calkins H, Judge D. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/ cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 2008; 5(5):258- 67. [ Links ]
9. Ahmad F. The molecular genetics of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Clin Invest Med. 2003; 26:167-78. [ Links ]
10. Kies P, Bootsma M, Bax J, Schalij M, van der Wall E. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Screening, diagnosis, and treatment. Heart Rhythm. 2006; 3(2): 225-34. [ Links ]
11. Bowles N, Ni J, Marcus F, Towbin J. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2002; 39(5): 892-5. [ Links ]
12. Van der Harst J, Suurmeijer A, van Veldhuisen D. Arrhythmogenic right ventricular cardiomyopathy: different manifestations as precursors of sudden death which might be prevented. Ned Tijdschr Geneesk. 2004; 149: 2396 -2402. [ Links ]
13. Peters S. Advances in the diagnostic management of arrhythmogenic right ventricular dysplasia cardiomyopathy. Int J Cardiol. 2006; 113(1): 4 - 11. [ Links ]
14. Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G, et al. Arrhythmogenic right ventricular dysplasia/ cardiomyopathy: need for an international registry. European Society of Cardiology and the Scientific Council on Cardiomyopathies of the World Heart Federation. J Cardiovasc Electrophysiol. 2000; 11(7): 827-32. [ Links ]
15. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom- Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Diseases of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994; 71(3): 215-218. [ Links ]
16. Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002; 40(8): 1445-50. [ Links ]
17. Yoerger DM, Marcus F, Sherrill D, Calkins H, Taubin JA, Zareba W, et al. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia. J Am Coll Cardiol. 2005; 45(6): 860-865. [ Links ]
18. Midiri M, Finazzo M. MR imaging of arrhythmogenic right ventricular dysplasia. Int J Card Imaging. 2001; 17: 297-304. [ Links ]
19. Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart. 2000; 83: 588-95. [ Links ]
20. Fontaine G, Fontaliran F, Hebert JL, Chemla D, Zenati O, Le carpentier Y, et al. Arrhythmogenic right ventricular dysplasia. Annu Rev Med.1999; 50: 17-35. [ Links ]
21. Santangelli P, Dello Russo A, Pieroni M, Casella M, Di Biase L, Burkrhardt JD, et al. Fragmented and delayed electrocardiograms within fibrofatty scar predict arrhythmic events in arryhtmogenic right ventricular cardiomyopathy: results from a prospective risk stratification study. Heart Rhythm. 2012; 9(8): 1200-6. [ Links ]
22. Uribe W. Guías Colombianas de Cardiología: Arritmias Ventriculares y Muerte Súbita. Rev Col Cardiol. 2011; 18 (s1): 63-70. [ Links ]
23. Marcus G, Glidden D, Polonsky B, Zareba W, Smith L, Cannom D, et al. Efficacy of Antiarrhythmic Drugs in Arrhythmogenic Right Ventricular Cardiomyopathy. J Am Coll Cardiol. 2009; 54(7): 609-15. [ Links ]
