










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicas UIS
versión impresa ISSN 0121-0319
Medicas UIS vol.28 no.1 Bicaramanga ene./abr. 2015
Hiperplasia suprarrenal congénita: origen de
trastornos del desarrollo y diferenciación sexual
Ana Herrera-Gómez*
*Estudiante de Medicina IX nivel. Universidad Industrial de Santander. Bucaramanga. Santander. Colombia.
Correspondencia: Srta. Ana Herrera. Calle 18 # 13-25. Barrio Gaitán. Bucaramanga. Santander. Colombia. Código postal: 680011. Correo electrónico: ana7hg@gmail.com
Artículo recibido el 15 de agosto de 2014 y aceptado para publicación el 20 de diciembre de 2014.
RESUMEN
La hiperplasia suprarrenal congénita es una patología de origen genético que se desarrolla por un déficit enzimático secundario a alteraciones en la síntesis de proteínas. Dicho déficit perturba las vías del metabolismo suprarrenal, específicamente las que ocurren en la corteza de la glándula, es decir, el metabolismo glucocorticoide, mineralocorticoide y de hormonas sexuales. Estas variaciones moleculares traducidas a la clínica se expresan en una variedad de presentaciones posibles, cada una dependiente de la enzima que se encuentre afectada, desde alteraciones electrolíticas, metabólicas, renales o del tracto gastrointestinal, pero hay una que es comúnmente encontrada, trastornos de desarrollo y diferenciación sexual. Dentro de las formas más severas de hiperplasia suprarrenal congénita se observan alteraciones de los caracteres sexuales externos que básicamente traducen en virilización de los genitales durante el desarrollo gestacional, así como también pubertad precoz, amenorreas, infertilidad, entre otros. MÉD.UIS. 2015;28(1):125-132.
Palabras Clave: Hiperplasia Suprarrenal Congénita. Desarrollo Sexual. Genética. Diferenciación Sexual. Caracteres Sexuales.
Adrenal hyperplasia congenital: beginning of developmental and sexual differentiation
disorders
ABSTRACT
Adrenal hyperplasia congenital is a genetic disease that develops from an enzyme deficiency secondary to alterations in protein synthesis. This deficit disturbs the pathways of adrenal metabolism, specifically occurring in the cortex of the gland, such as, the glucocorticoid, mineralocorticoid and the sex hormones metabolism. These molecular variations translated to the clinics are expressed in a variety of possible presentations, each depending of the enzyme that is affected, since electrolyte, metabolic, renal abnormalities or and gastrointestinal tract disturbances, but one of these is commonly for all presentation, the development and sexual differentiation disorders. Among the most severe forms of adrenal hyperplasia congenital alterations of the external sexual characters that basically translate into virilization of the genitals duringgestational development, as well as precocious puberty, amenorrhea, infertility observed, among other. MÉD.UIS. 2015;28(1):125-132.
Keywords: Adrenal Hyperplasia congenital. Sexual Development. Genetics. Sex Differentiation. Sex Characteristics.
¿Cómo citar este artículo?: Herrera-Gómez A. Hiperplasia suprarrenal congénita: origen de
trastornos del desarrollo y diferenciación sexual. MÉD.UIS. 2015;28(1):125-132.
INTRODUCCIÓN
La Hiperplasia Suprarrenal Congénita (HSC) es un trastorno autosómico recesivo con una frecuencia de presentación relativamente baja, según datos estadísticos a nivel mundial la incidencia de la enfermedad puede variar desde de 1 en 10 000 a 18 000 personas1. Actualmente, en Colombia no se cuenta con estudios epidemiológicos o estadísticos de incidencia o prevalencia de la enfermedad1. La HSC se caracteriza por déficit de enzimas que participan en el metabolismo en la corteza de la glándula suprarrenal. Así como existe un espectro de enzimas que pueden estar deficientes en la enfermedad hay una amplia gama de formas clínicas que se pueden expresar, que van desde signos leves y asintomáticos hasta comprometer seriamente la vida del paciente.
Se conoce que las principales funciones metabólicas de la corteza suprarrenal engloban las vías mineralocorticoide, glucocorticoide y hormonal sexual. En la HSC puede existir desde una afección leve de alguna de las vías, hasta un compromiso severo de los tres metabolismos mencionados, pero en la mayoría de las formas clínicas expresadas se encuentra cierto grado de compromiso del metabolismo de hormonas sexuales, de este modo, en pacientes masculinos la afectación no es tan severa; sin embargo, el estímulo androgénico en pacientes femeninos se expresa con cambios importantes a nivel del desarrollo genital externo denominado trastorno del desarrollo sexual 46XX. Así pues la siguiente revisión de tema tiene como objetivo tratar las generalidades de la HSC, enfocado en el trastorno del desarrollo y diferenciación sexual femenino con el fin de reflexionar sobre los posibles caminos a tomar en un futuro para el abordaje de dicha enfermedad.
HIPERPLASIA SUPRARRENAL CONGÉNITA: ORIGEN DE TRASTORNOS DEL DESARROLLO Y DIFERENCIACIÓN SEXUAL
La HSC es una patología de origen genético, de carácter autosómica recesiva, asociado a un déficit enzimático en la vía de la esteroidogénesis2. En el metabolismo de la glándula suprarrenal se obtienen varios compuestos hormonales como catecolaminas, mineralocorticoides, glucocorticoides y hormonas sexuales, siendo estos tres últimos los afectados en la HSC. A grandes rasgos la vía mineralocorticoide está encargada de regular la excreción renal, la vía glucocorticoide actua en situaciones de ayuno o estrés aumentando la gluconeogénesis y las hormonas sexuales se encargan de procesos de diferenciación sexual en etapa perinatal y maduración gonadal durante la pubertad3,4.
En la HSC hay alteración de la producción de aldosterona, cortisol y andrógenos, que son los compuestos más representativos del metabolismo de la corteza suprarrenal5. No obstante el cuerpo humano está apto para detectar y responder dichas bajas hormonales activando el eje hipotálamohipofisis- adrenal gracias a un proceso de retroalimentación4. Fisiológicamente el cortisol hace retroalimentación negativa en el hipotálamo y la hipófisis disminuyendo o aumentando la producción de hormona liberadora de corticotropina y hormona adrenocorticotropa respectivamente, que actúan finalmente en la glándula suprarrenal activando las vías metabólicas de la corteza suprarrenal6.
En la HSC existe el déficit de cinco enzimas principalmente; la 21-hidroxilasa (21 OH), 11 β hidroxilasa (11β OH), 17 α hidroxilasa (17α OH), 3 β hidroxiesteroide deshidrogenasa (3β eDOH) y StAR (proteína reguladora de las esteroidogénesis aguda), siendo la 21-hidroxilasa las más común en 95% de los casos7. En la figura 1 se presentan las vías de metabolismo de la corteza suprarrenal, señalando que todas en común inician con el colesterol como precursor de los compuestos a obtener y se indica cada una de las enzimas anteriormente mencionadas que actúan en la fisiopatología de la HSC. Debido al déficit enzimático ocurre una interrupción en la producción de cortisol, que envía señales a hipotálamo e hipófisis aumentado la producción de hormona liberadora de corticotropina y adrenocorticotropa, lo que hiperestimula la corteza suprarrenal; sin embargo, ya que la vía está detenida por la pérdida enzimática hay aumento del metabolito previo a la conversión originando lo que se conoce como hiperplasia suprarrenal congénita, que dependiendo de la enzima y momento de la síntesis interrumpida, se ira a desarrollar una forma clínica de la patología2.
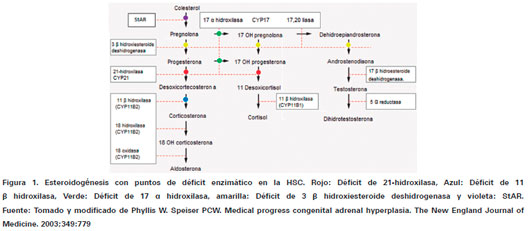
FORMAS CLÍNICAS
Déficit de 21-hidroxilasa
Es la presentación más común HSC, en la que se detiene el paso de progesterona a desoxicorticosterona para finalizar en aldosterona y el paso de 17 OH-progesterona a desoxicortisol para finalizar en cortisol2 (Ver Figura 1. Señalización roja), lo que lleva a que dichas hormonas represadas tengan que metabolizarse por la única vía alterna y convertirse en andrógenos, aumentado considerablemente sus niveles sanguíneos1,3. En general la HSC se muestra en dos maneras clínicas; clásica y no clásica8: a) Forma clásica: se presenta con una incidencia de 1 en 10 000 a 15 000 personas, esta forma de HSC es también llamada severa y se subclasifica en virilizante simple y perdedora de sal, con una prevalencia de presentación de 70% y 30% de los casos respectivamente7,8,9.
La forma virilizante simple presenta niveles residuales de la enzima deficiente por lo cual hay concentraciones mínimas de cortisol y aldosterona que sí llegan a producirse. En estos pacientes la clínica expresada se da por efecto del aumento de hormonas sexuales como procesos de virilización y pubertad precoz mientras que los efectos clínicos en el metabolismo renal y de la glucosa son subsanados con la poca producción existente2,10.
La forma perdedora de sal tiene comprometida en su totalidad la producción de aldosterona y cortisol, por tanto, los efectos glucocorticoides, mineralocorticoides y sexuales del metabolismo suprarrenal son más severos. Las manifestaciones de carácter sexual aparecen desde el periodo perinatal con hallazgos como virilización de genitales en fetos femeninos secundario al aumento de hormonas sexuales, mientras que en fetos masculinos la virilización ocurre de manera natural y la sospecha o manifestaciones clínicas se darán en etapas de la niñez y pubertad del paciente3. En la vía mineralocorticoide y glucocorticoide la carencia de aldosterona y cortisol se expresa durante el periodo neonatal con alteraciones metabólicas que varían desde pérdida de sal, alteración en la función cardiaca, acidosis metabólica y estados de hiperpotasemia, hiponatremia e hipoglucemia severa expresada en síntomas como inapetencia, vómito, diarrea y deshidratación en el recién nacido11,12. En la forma perdedora de sal es necesario tratamiento médico para evitar secuelas en el desarrollo neurológico del paciente1.
b) Forma no clásica: esta presentación de HSC tiene una incidencia de 1 en 1000 personas. En estos casos los pacientes tienen la alteración metabólica pero son asintomáticos o con síntomas leves, predominantemente asociados a la estimulación hormonal sexual8,9,12. La clínica es más tardía y variable que la forma clásica, por ejemplo durante la niñez femenina se asocia a pubertad precoz, crecimiento acelerado que repercutirá en la talla adulta, donde se observa desde hipertrofia del clítoris, hirsutismo, alteraciones del ciclo menstrual, síndrome de ovario poliquístico hasta infertilidad, en tanto que el hombre puede presentar una pubertad precoz, oligoespermia, infertilidad y alteración testicular como no funcionalidad ni crecimiento de los mismos, pero la mayoría cursa asintomático8,9,13.
Déficit de 11 β hidroxilasa
Es el segundo déficit enzimático de mayor frecuencia, con una incidencia de 1 por cada 100 000 nacimientos, que traduce en 3% a 5% de los casos de HSC1,13. Esta enzima se encarga de la conversión de desoxicorticosterona a corticosterona y de desoxicortisol a cortisol (Ver Figura 1. Señalización azul), y al igual que en el déficit de 21-hidroxilasa, se produce un aumento de los metabolitos previos a la acción de esta enzima y un desplazamiento hacia la conversión de andrógenos1.
a) Forma clásica: en el déficit de la 11 β hidroxilasa no existe la subclasificación de perdida salina, debido a que los metabolitos alcanzados en la vía de la aldoterona son suficientes para no producir sintomatología. Sin embargo los signos clínicos expresados como virilización de genitales externos en mujeres es similar a la clínica expresada por el déficit de la 21-hidroxilasa, mencionado anteriormente13.
b) Forma no clásica: es infrecuente y generalmente cursa asintomática, y cuando se expresa clínicamente lo hace con leves cambios por estimulación hormonal sexual como pubertad precoz, hirsutismo, alteraciones del ciclo menstrual o síndrome de ovario poliquístico7,14.
Déficit de 17 α hidroxilasa
En esta deficiencia se ve inactiva la conversión de pregnolona a 17 hidroxipregnenolona y de progesterona en 17 hidroxiprogesterona (Ver Figura 1. Señalización verde). En esta forma de presentación se alteran las vías glucocorticoide y hormonal sexual es decir se inhabilita la producción de cortisol y testosterona mientras que la vía de la aldosterona no sufre modificaciones, es por eso que clínicamente la aldosterona produce inhibición del sistema reninaangiotensina, reteniendo sodio y con él líquidos que aumentan las cifras tensiónales1,13. Esta forma clínica tiene una presentación diferente a las anteriormente mencionadas, sus manifestaciones pueden ser agrupadas por género de la siguiente manera:
a) Femenino: No se presenta virilización de genitales si no alteraciones en edades más avanzadas como ausencia de pubarquia.
b)Masculino: Existe alteración genital dependiente del grado de déficit enzimático, puede alcanzar un trastorno del desarrollo sexual 46XY debido a que en el déficit de la 17 α hidroxilasa las vías metabólicas no llegan a la producción de testosterona, pues solo esta activa la vía mineralocorticoide1,13.
1.4 Déficit de 3 β hidroxiesteroide deshidrogenasa
Esta deficiencia enzimática origina represamiento de los metabolitos en la conversión de pregnenolona a progesterona, de 17 OH-pregnenolona a 17 OHprogesterona y de deshidroepiandrosterona a androstenodiona (Ver Figura 1. Señalización amarilla). Es decir involucra las tres vías de metabolismo de la corteza suprarrenal: mineralocorticoide, glucocorticoide y androgénica1. Esta presentación es poco frecuente y la clínica incluye las presentaciones clásicas y no clásicas:
a) Forma clásica: es una presentación clínica severa; en la vía mineralocorticoide se ve expresada la clínica de perdedora de sal mencionada anteriormente, y en la vía androgénica se expresa en hombres con una masculinización deficiente con hallazgos de micropenes o alteraciones como hipospadias, y en género femenino hay exceso de dehidroepidandrosterona expresada como virilización intrauterina13.
b) Forma no clásica: es una presentación clínica leve, expresando preponderantemente la alteración sexual y en menor medida la afección mineralocorticoide y glucorticoide, clínicamente los pacientes masculinos presentan hipogonadismo y las pacientes femeninas presentan en edades avanzadas alteración del ciclo menstrual, hirsutismo o síndrome de ovario poliquístico1,15.
1.5 Déficit de proteína StAR (proteína reguladora de las esteroidogénesis aguda)
Este déficit es también conocido como hiperplasia lipoidea congénita, es una presentación poco frecuente y severa, se han descrito tan solo cerca de 100 casos13. La proteína StAR (Ver Figura 1. Señalización violeta) se encarga del paso de colesterol desde el citoplasma a la mitocondria donde continúan las vías metabólicas, por esta razón el colesterol no se convierte en pregnenolona para iniciar con la esteroidogenesis14. La acumulación de colesterol en el citoplasma celular se vuelve tóxica lo que lleva a la muerte de estas células y a una insuficiencia suprarrenal total13. Debido a que no puede existir ningún precursor en las vías mineralocorticoide, glucocorticoide ni hormonal sexual el déficit es generalizado y la clínica se expresa con la depleción de todos estos metabolismos:
a) Mineralocorticoide: déficit de aldosterona, se expresa con hipotensión, hiponatremia y perdida de sal13.
b) Glucocorticoide: déficit de cortisol, se evidencia por hipoglicemia, disfunción cardiaca y vascular12
c) Hormonal sexual: en recién nacidos varones se presenta trastornos del desarrollo y diferenciación sexual ya que no se está produciendo andrógenos mientras que en pacientes femeninas los caracteres sexuales externos son normales13,14.
GENÉTICA DE LA HSC
La HSC se encuentra en el grupo enfermedades genéticas autosómicas recesivas, donde ocurren alteraciones en los genes que codifican las enzimas encargadas de la esteroidogénesis14. Los genes que transcriben posteriormente a la enzima 21 hidroxilasa, cuyo déficit es el más común en HSC, son CYP21A que es el gen activo y el pseudogén CYP21P, ambos se localizan en el extremo 3' del brazo corto del cromosoma 6 (6p21.3)3,5,16. Si se compara el gen activo y el seudogén CYP21 presentan 96% de homología3.
En la figura 2 se observa los tipos de lesiones genéticas que ocurren en la HSC, el 75% de ellas suceden por mutaciones puntuales (Ver Figura 2. Dibujo A) se subdivide en:
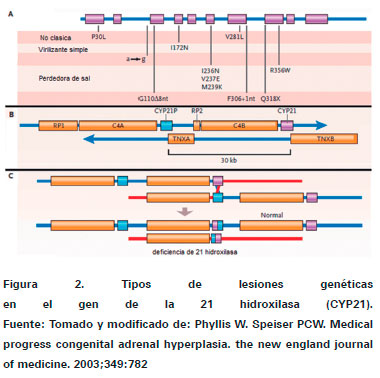
- Asociada a forma no clásica: p.Pro30Leu, p.Val281Leu y p.Pro453Ser .
- Asociada a forma virilizante simple: p.Ile172Asn y p.Arg426His .
- Asociada a forma perdida salina: p.Ile236Asn, p.Val237Asp, p.Met239Lys, p.Val281Leu, c.923 dupT, p.Gln318Stop y p.Arg356Trp2,12 .
También ocurren deleciones en el 15% de la veces (Ver Figura 2, Dibujo B) donde se ve un segmento cromosómico con genes normales identificando los CYP21 activo y el seudogén y otro segmento cromosómico con una deleción de 30 kb de longitud con desaparición del gen de la 21 hidroxilasa produciendo la patología, en el dibujo C se observa una lesión genética debido a recombinación desigual en la meiosis, también pueden ocurrir grandes conversiones (9%) y duplicaciones (2%)3,12.
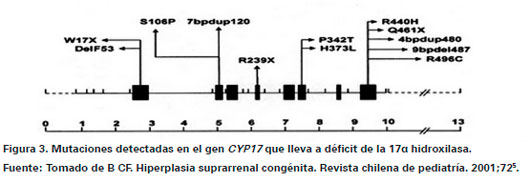
La enzima 11β-hidroxilasa es codificada del gen CYP11B1, localizado en el cromosoma 8 en la parte media del brazo largo1. Entre las mutaciones descritas la más frecuente es la R448H, que destruyen a actividad enzimática produciendo la HSC15. La deficiencia de 17α hidroxilasa está determinada por defectos en el gen CYP17, entre las diferentes mutaciones descritas la mayoría comprometen el exón 8 del gen que puede llevar a inactividad total o parcial de la enzima14. En la Figura 3 se observa más mutaciones para el gen CYP17.
El gen que codifica la 3 β hidroxiesteroide deshidrogenasa es el 3βHSD, el cuerpo humano tiene dos tipos de este gen que codifica para isoenzimas con 30% de homología, uno de ellos es el 3β1HSD que se expresa en placenta y tejidos periféricos y el 3β2HSD que se expresa a nivel suprarrenal y gonadal la cual es clave para la HSC15. Ambos genes se localizan en el brazo corto del cromosoma 1 en la región 11 a 131,15.
Las alteraciones genéticas involucradas con la proteína StAR relacionan tres mutaciones específicas, la R193X, la Q258X y mutación que altera el splicing en el exón 5 del gen lo que no permite la correcta codificación y formación de la proteína impidiendo su correcto funcionamiento y consiguiente aparición de la patología14.
TRASTORNO DEL DESARROLLO Y DIFERENCIACIÓN SEXUAL 46XX
Principios de diferenciación sexual
Embriológicamente la determinación sexual está a cargo de señales genéticas y hormonales diferenciadas en tres etapas: la genética, la gonadal y la fenotípica17.
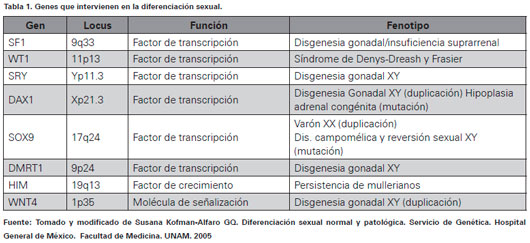
La etapa genética ocurre en la fecundación con la unión del óvulo y el espermatozoide; el óvulo (X/X) genéticamente aporta el cromosoma X y el espermatozoide (X/Y) puede aportar tanto cromosoma X, para formar feto femenino, o aportar cromosoma Y para crear un feto masculino18. Esta gran diferencia dada por el cromosoma Y está sustentada por los genes que en él alberga, se encuentra el gen SRY, ubicado en el brazo corto del cromosoma Y (Yp11.3), que codifica el factor determinante testicular estableciendo el desarrollo de gónadas masculinas, que al no expresarse en fetos XX ordena el desarrollo gonadal femenino dando el inicio a la etapa gonadal19,20. Además del SRY existen otros genes que actúan de manera más temprana y hacen parte de todo el proceso de diferenciación sexual, entre ellos se encuentra WT1 (Tumor de Willms 1) localizado en 11p13 y SF1 (Factor eseteroidogénico 1) ubicado en 9q33, ambos actúan en el mantenimiento de la cresta urogenital20. En la Tabla 1 se resumen los genes que actúan en la diferenciación sexual y sus características.
En la etapa fenotípica el proceso embriológico ocurre de manera igual para ambos sexos hasta la semana séptima del embarazo cuando inicia una producción de proteínas y cambios celulares denominado determinación sexual, en estas primeras etapas la cresta urogenital se subdivideen cresta urinaria y cresta gonadal, siendo esbozo de lo que posteriormente son las gónadas. En el mesonefros se encuentran los conductos de Wolff y en la cresta gonadal se origina los conductos de Müller que se convertirán en los genitales internos. En cuanto al desarrollo de gentiles externos se hace una división de la membrana cloacal en membrana urogenital y anal. De la membrana urogenital se constituirá en tubérculo genital, pliegues urogenitales y pliegues labioescrotales21. Tanto la diferenciación sexual interna y externa dependede estímulo hormonal, luego de que ocurre el proceso embrionario los genitales externos pueden diferenciarse en sentido masculino si recibe estimulo hormonal de dihidrotestosterona, convirtiendo el tubérculo genital en pene y los repliegues labioescrotales en bolsas escrotales. Mientras que en fetos femeninos no ocurre esta estimulación por lo cual el tubérculo genital se convierte en clítoris y los pliegues urogenitales forman los labios menores y los repliegues labioescrotales forman labios mayores18,21.
HSC asociada al trastorno de desarrollo sexual 46XX (46 XX DSD)
El trastorno de desarrollo sexual 46 XX, inicialmente conocido como pseudohermafroditismo femenino, hace parte de un grupo de alteraciones denominadas Trastornos del Desarrollo Sexual (DSD), anteriormente nombrados intersexo. Donde también se encuentran el trastorno de desarrollo sexual 46XY (llamado pseudohermafroditismo masculino), trastorno de desarrollo sexual ovotesticular, llamado hermafroditismo verdadero, trastorno de desarrollo sexual 46 XX testicular, llamado sexo reverso XX, y disgenesia gonadal completa 46 XY ,llamado sexo reverso XY22-24.
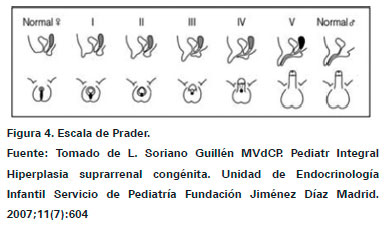
El trastorno de desarrollo sexual 46 XX se encuentra asociado a la hiperplasia suprarrenal congénita, según la enzima y el grado de compromiso en la HSC las pacientes femeninas pueden presentar esta característica. Genéticamente y gonadalmente el proceso de diferenciación sexual ocurre correctamente, sin embargo, producto de la hiperestimulación suprarrenal y el aumento de andrógenos por la HSC se produce un estímulo hormonal que compromete el desarrollo genital externo21,25. Durante el embarazo si un feto femenino presenta HSC tiene un aumento de la estimulación androgénica, como se mencionó, dependiendo del compromiso suprarrenal puede existir un estímulo androgénico mayor o menos en las diferentes formas de HSC, es por esto que la ambigüedad de los genitales externos puede variar, actualmente se utiliza la escala de Prader para catalogar el grado de compromiso a nivel genital, que puede variar desde una clitoromegalia ligera (Estadios I-II) y fusión posterior hasta fusión labioescrotal completa y seno urogenital (Estadio V) (Ver Figura 4)13,23.
Acción prenatal
Si se realiza un diagnóstico durante el embarazo, la acción farmacológica y tratamiento ayudan disminuir los síntomas al momento del nacimiento, y si se asume que las formas severas llegan a ser mortales para los neonatos, el riesgo-beneficio en la detección temprana de la enfermedad se hace imprescindible. Según las guías de la sociedad de endocrinología, actualmente está indicada la realización de exámenes prenatales con el fin de realizar diagnósticos oportunos que reduzcan morbilidad neonatal, sin embargo estos deben realizarse bajo atención especializada en centros médicos de alto nivel y por ahora el tratamiento prenatal aun continua bajo estudios experimentales25.
Acción postnatal
El abordaje global para la HSC en un recién nacido esta guiado en detener los síntomas, dependiendo de la forma clínica expresada, pueden ser síntomas que requieran hospitalización y tratamiento inmediato desde el nacimiento o puede expresar clínica en edades mayores como durante la pubertad. Cualquiera que sea la edad del paciente, la atención médica se debe otorgar de parte de un grupo multidisciplinario que contemple la consulta de pediatra, genetista y endocrinólogo dentro del grupo.
Adicional al tratamiento dado para alteraciones metabólicas, los trastornos de desarrollo sexual en pacientes femeninas necesitan de un abordaje adicional y personalizado, sustentado en el hecho que aunque se equilibre la producción suprarrenal con medicamentos y suplementos hormonales, la progresión de la virilización durante el periodo neonatal no tienen regresión, por lo cual, es necesario pensar posibilidades quirúrgicas reconstructivas. De igual manera esta decisión debe ser tomada por un grupo interdisciplinario que evalúe el caso en particular1,26.
CONCLUSIONES
La hiperplasia suprarrenal congénita hace parte de patologías de origen genético asociada a mutaciones en los genes que codifican para proteínas, cuya función es catalizar procesos metabólicos a nivel suprarrenal. Es una enfermedad ampliamente estudiada cuya forma más frecuente, la del déficit de 21-hidroxilasa, presenta formas clínicas en las cuales varia el espectro de presentación de severidad. Así como puede haber pacientes seriamente afectados en el metabolismo renal y energético que requieren de una asistencia médica inmediata desde el nacimiento para evitar secuelas, pueden existir presentaciones de la patología casi asintomáticas, en ocasiones levemente expresadas en la adolescencia que hacen sospechar la enfermedad y cuyo tratamiento es igualmente sencillo.
Si bien la ciencia médica y biológica está encaminada en el estudio profundo de enfermedades como la hiperplasia suprarrenal congénita, estas patologías de origen genético son de complicado abordaje. Hoy por hoy, existen métodos de estudios prenatales con el fin de realizar detección oportuna de la enfermedad antes del nacimiento y preparar la atención del neonato.
Sin embargo, en la actualidad no existe un tratamiento estándar aprobado durante el embarazo, aunado al hecho de que en países como el nuestro la captación de pacientes por medio de controles prenatales se realiza en etapas tardías de la gestación, en la mayoría de los casos. El abordaje de pacientes con hiperplasia suprarrenal congénita se realiza posterior al nacimiento examinando directamente el recién nacido haciendo un estudio más certero de las condiciones del paciente, mediante un grupo multidisciplinario que garantice un manejo integral. Se hace necesario que la medicina continúe con la investigación de enfermedades genéticas como la hiperplasia suprarrenal congénita, con el fin encontrar nuevas técnicas de detección y métodos seguros de tratamiento para los pacientes afectados, de esta manera se disminuiría las tasas de morbi-mortalidad, costos en estancia hospitalaria y secuelas, otorgando una mejor calidad de vida para los paciente y las familias afectadas por la enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1. Sepúlveda J, Sanguino L, Jaimes H. Hiperplasia adrenal congénita. Rev colomb obstet ginecol. 2001;52(4). [ Links ]
2. Milagros A, Begoña E. Hiperplasia suprarrenal congénita no clásica o tardía. Rev Esp Endocrinol Pediatr. 2012;3(Suppl):61-73. [ Links ]
3. Fonseca D, Gutiérrez A, Silva C, Coll M, Malo G, Orjuela C, et al. Identificación de mutaciones puntuales del gen de la 21-hidroxilasa en pacientes afectados con hiperplasia suprarrenal congénita. Biomédica(Bogotá). 2005;25(2):220-30. [ Links ]
4. Rodríguez JM. El papel del receptor de glucocorticoide en el estrés temprano. Univ. Méd. Bogotá (Colombia). 2010;51(4):385-91. [ Links ]
5. Rege J, Rainey WE. The steroid metabolome of adrenarche. J Endocrinol. 2012;214(2):133-43. Epub 2012 Jun 19. [ Links ]
6. Navas C, Zapata D. Aspectos inmunológicos en la depresión. Rev venez oncol. 2009;21(4):244-52. [ Links ]
7. Angoorani H, Haratian Z, Halabchi F. Congenital Adrenal Hyperplasia in an Elite Female Soccer Player; What Sports Medicine Clinicians Should Know about This?. Asian J Sports Med. 2012;3(3):209-13. [ Links ]
8. Reisch N, Arlt W, Krone N. Health Problems in Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency. Horm Res Paediatr. 2011;76(2):73-85 Epub 2011 May 18. [ Links ]
9. Cheviakoff S, Youlton R. Sindrome de hiperplasia adrenal congenita no clasica y embarazo. Rev chil obstet ginecol. 2003;68(1):28-31. [ Links ]
10. Al-Maghribi H. Congenital adrenal hyperplasia: problems with developmental anomalies of the external genitalia and sex assignment. Saudi J Kidney Dis Transpl. 2007;18(3):405-13. [ Links ]
11. Martínez MA, Hernández JI, Ramírez CA, Cordova LP, Esparza- Ledesma HM. Hiperplasia suprarrenal congénita secundaria a deficiencia de 21-hidroxilasa: Reporte de un caso. Bol Clín Hops Infant Edo Son. 2007;24(1):38-41. [ Links ]
12. Speiser PW, White PC. Medical progress congenital adrenal hyperplasia. N Engl J Med. 2003;349:776-88. [ Links ]
13. Soriano L, Velázquez M. Hiperplasia suprarrenal congénita. Unidad de Endocrinología Infantil. Servicio de Pediatría. Fundación Jiménez Díaz, Madrid. 2007;11(7):601-10. [ Links ]
14. Fardella C. Hiperplasia suprarrenal congénita. Rev. chil. pediatr. 2001;72(5):408-15. [ Links ]
15. Lusa LG, Lemos-Marini SH, Soardi FC, Ferraz LF, Guerra-Júnior G, Mello MP. Structural aspects of the p.P222Q homozygous mutation of HSD3B2 gene in a patient with congenital adrenal hyperplasia. Arq Bras Endocrinol Metab. 2010;54(8):768-74. [ Links ]
16. Carvajal F, Montesino T, Espinosa T, Navarrete J, Pérez C. Forma no clásica de hiperplasia adrenal congénita en la niñez y adolescencia. Rev Cubana Endocrinol. 2010;21(1):62-73. [ Links ]
17. Kofman-Alfaro S, Queipo G. Diferenciación sexual normal y patológica. Mensaje Bioquímico. 2005;29:109-18. [ Links ]
18. MacLaughlin DT, Donahoe PK. Sex Determination and Differentiation. N Engl J Med. 2004;350(4):367-78. [ Links ]
19. Oliva R. Genes del cromosoma Y. Significado clínico. En: SIMPOSI SOBRE INFERTILITAT MASCULINA: GENÈTICA I AMBIENT: Segona edició de sabadell universitat estiu; 2003. [ Links ]
20. Öçal G. Current Concepts in Disorders of Sexual Development. Journal Clin Res Pediatr Endocrinol. 2011;3(3):105-14. [ Links ]
21. Rey R. Diferenciación sexual embrio-fetal: De las moléculas a la anatomía. Rev chil anat. 2001;19(1):75-82. [ Links ]
22. Lee PA, Houk CP, Ahmed SF, Hughes IA; International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus Statement on Management of Intersex Disorders. Pediatrics. 2006;118(2):e488-500. [ Links ]
23. Erdoğan S, Kara C, Uçaktürk A, Aydin M. Etiological Classification and Clinical Assessment of Children and Adolescents with Disorders of Sex Development. J Clin Res in Pediatr Endocrinol. 2011;3(2):77-83. Epub 2011 Aug 6. [ Links ]
24. Kousta E, Papathanasiou A, Skordis N. Sex determination and disorders of sex development according to the revised nomenclature and classification in 46,XX individuals. Hormones (Athens). 2010;9(3):218-31. [ Links ]
25. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al.; Endocrine Society. Congenital Adrenal Hyperplasia Due to Steroid 21-hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2010;95(9):4133-60. [ Links ]
26. Islas LP, Jiménez JR, Verduzco M. Pseudohermafroditismo femenino por hiperplasia suprarreanl congénita. Rev mex pediatr. 2005;72(2):74-7. [ Links ]
