










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkMedicas UIS
Print version ISSN 0121-0319
Medicas UIS vol.29 no.2 Bucaramanga May/Aug. 2016
https://doi.org/10.18273/revmed.v29n2-2016011
DOI: http://dx.doi.org/10.18273/revmed.v29n2-2016011
Hibridación genómica comparativa: su
interpretación y uso como herramienta
diagnóstica en retardo mental inespecífico y
síndromes de microdeleción/microduplicación
Stephania Posada Guiran*
Victoria Andrea Osorio M**
Lizeth Gabriela Garzón Guerrón*
Julián Ramírez-Cheyne***
* Estudiante VIII semestre de Medicina. Facultad Ciencias de la Salud. Universidad Libre. Cali. Colombia.
** Estudiante VII semestre de Medicina. Facultad Ciencias de la Salud. Universidad Libre. Cali. Colombia.
*** Médico. Profesor Universidad del Valle. Profesor Universidad Libre. Cali. Colombia.
Correspondencia: Dr. Julián Ramiréz Cheyne. Dirección: Calle 4B # 36-00. Universidad del Valle, Sede San Fernando. Edificio 116. Departamento de Morfología. Laboratorio de Citogenética. Cali. Colombia. Correo electrónico: juracheyne@gmail.com
Artículo recibido el 27 agosto de 2015 y aceptado para publicación el 16 de abril de 2016.
RESUMEN
En Colombia el cariotipo por bandeo es todavía la prueba inicial más usada en el estudio de pacientes con retardo mental o con anomalías congénita múltiples. Sin embargo, las técnicas moleculares, particularmente la hibridación genómica comparativa con microarrays, han permitido identificar un número creciente de síndromes de microduplicación o microdeleción en estos pacientes, de modo que mundialmente esta es hoy en día la tecnología de elección para evaluar alteraciones del número de copias. Se realiza esta revisión de la literatura con el objetivo de brindar al personal médico información actualizada acerca de la interpretación y el papel de la hibridación genómica comparativa con microarrays como herramienta diagnóstica en retardo mental inespecífico, síndromes de microdeleción/ microduplicación y análisis cromosómico prenatal. MÉD.UIS. 2016;29(2):137-44.
Palabras clave: Hibridación Genómica Comparativa. Discapacidad intelectual. Deleción Cromosómica. Duplicación Cromosómica. Diagnóstico Prenatal.
Comparative genomic hybridization: its interpretation and use as a diagnostic tool in
nonspecific mental retardation and microdeletion/microduplication syndromes
ABSTRACT
In Colombia the banding karyotype is still the initial test used in the study of patients with mental retardation or with multiple congenital anomalies. However, molecular techniques, particularly comparative genomic hybridization with microarray have identified a growing number of microdeletion or microduplication syndromes in these patients, so that globally this is the technology of choice nowaday for evaluating copy number alterations. This literature review is aimed at providing to medical personnel the latest information regarding the interpretation and role of comparative genomic hybridization as a diagnostic tool in nonspecific mental retardation, microdeletion/ microduplication syndromes and prenatal chromosomal analysis. MÉD.UIS. 2016;29(2):137-44.
Keywords: Comparative Genomic Hybridization. Intellectual Disability. Chromosome Deletion. Chromosome Duplication. Prenatal Diagnosis.
¿Cómo citar este artículo?: Posada S, Osorio VA, Garzón LG, Ramírez-Cheyne J. Hibridación genómica comparativa: su interpretación y uso como herramienta diagnóstica en retardo mental inespecífico y síndromes de microdeleción/microduplicación. MÉD.UIS. 2016;29(2):137-44.
INTRODUCCIÓN
El retraso mental es un desorden clínico con gran impacto para la salud pública y la sociedad; sin embargo, su etiopatogenia es poco entendida. Se define como un daño significativo de las funciones cognitiva y adaptativa, con inicio antes de los 18 años de edad. Puede hacerse evidente durante la infancia o en la niñez temprana como retraso del desarrollo, pero es mejor diagnosticado durante los años escolares1. A nivel mundial se ha estimado una prevalencia aproximada del uno al cuatro porciento. En Latinoamérica la prevalencia puede ser cuatro veces mayor, por su asociación a factores como la desnutrición, las complicaciones obstétricas y perinatales, la prematurez, la intoxicación por plomo, las infecciones del sistema nervioso central y la pobreza2,3.
Las causas genéticas de discapacidad intelectual pueden estudiarse con diferentes técnicas. En Colombia, aunque la citogenética molecular se aplica desde los noventa4, el cariotipo por bandeo es todavía la prueba inicial más usada, y cuando este resulta normal, las pruebas siguientes incluyen citogenética molecular o genética molecular. No obstante, en los últimos cinco años, siguiendo la tendencia mundial, se ha venido incrementando en el país el uso de la Hibridación Genómica Comparativa con microarrays (aCGH, por sus siglas en inglés) como prueba inicial para estos casos5. En pacientes con Retardo Mental (RM) el uso de esta tecnología ha permitido la identificación de un número creciente de Síndromes de Microduplicación o Microdeleción (MMS, por sus siglas en ingles)6, cuya aparición se facilita por secuencias de ADN repetitivas que predisponen a recombinaciones homólogas desiguales y son responsables de los rearreglos cromosómicos recurrentes7-9.
El cariotipo con 550 bandas G permite la identificación de alteraciones numéricas, también llamadas aneuploidías, y del mayor número de alteraciones estructurales con tamaño mínimo de cinco megabases. La aplicación de las técnicas moleculares, como el aCGH, en el estudio cromosómico han permitido diagnosticar reorganizaciones crípticas por debajo de la resolución del microscopio óptico10,11.
Mundialmente, el aCGH se ha convertido en la tecnología de elección para evaluar alteraciones del número de copias, también llamadas Variaciones del Número de Copias (CNV, por sus siglas en inglés), asociadas a características como RM, retraso del desarrollo, autismo o dismorfismo12 . Es importante que los médicos se documenten sobre la utilidad, indicaciones y características operativas del aCGH, ya que debido a sus ventajas sobre el cariotipo y otras técnicas moleculares es probable que se consolide como la prueba de primera línea para el análisis cromosómico en los próximos años, incluso en el escenario del diagnóstico prenatal de fetos con dismorfismo13 . El objetivo de este artículo es realizar una revisión acerca de la interpretación y uso del aCGH como herramienta diagnóstica en RM inespecífico, MMS y análisis cromosómico prenatal haciendo una comparación con el cariotipo.
METODOLOGÍA DE BÚSQUEDA
Se realizó una búsqueda en las bases de datos PubMed- MEDLINE y Google Scholar, usando los términos MeSH "Comparative Genomic Hybridization"; "Intellectual Disability"; "Chromosome Deletion", "Chromosome Duplication"; "Prenatal Diagnosis". Se combinó el primer término con cada uno de los demás usando el operador booleano AND. Se incluyeron artículos publicados desde 1998.
HIBRIDACIÓN GENÓMICA COMPARATIVA CON MICROARREGLOS
TÉCNICA
En el aCGH, el ADN del paciente y un ADN control son marcados, mezclados en proporción 1:1 e hibridados junto con un ADN de un donante normal. Esta hibridación in situ con fluorescencia, hecha en placas con pozos de secuencias genómicas definidas, resulta en un color amarillo de las regiones no afectadas por CNV, mientras que los colores verdes y rojos indican pérdidas o ganancias del ADN del paciente respectivamente. Además se pueden detectar polimorfismos de nucleótidos que proveen información adicional sobre pérdida de heterocigosidad, indicando deleción o disomia uniparental. El aCGH es usado como método para revisar todo el genoma y la resolución depende del número y tamaño de las sondas usadas14-6.
INTERPRETACIÓN
Con los problemas técnicos resueltos, hoy el reto principal es la interpretación. La decisión más crítica es si una CNV es considerada benigna o patológica.
Para esto, se emplea un sistema de tres categorías para clasificar las CNV halladas en un aCGH: probablemente patógena, probablemente benigna o Variante de Significancia Desconocida (VUS, por sus siglas en inglés)16-18. La asignación de una CNV a una de estas categorías puede hacerse revisando la literatura y usando buscadores genómicos que muestran las CNV patológicas y benignas reportadas19; adicionalmente, muchos laboratorios mantienen sus propias bases de datos.
Si una CNV no ha sido reportada previamente, el proceso de clasificación involucra el análisis del tamaño, contenido y asociación con fenotipos anormales, teniendo varias consideraciones generales. Así, se clasifican como alteraciones candidatas para ser responsables de los trastornos o patológicas, aquellas que implican pérdidas, tienen gran tamaño, son ricas en genes o son de novo; se clasifican como alteraciones candidatas para no ser responsables de los trastornos o benignas aquellas que no contienen genes, contienen genes que no se ha demostrado que sean sensibles a la dosis, las cercanas a otras alteraciones reportadas como benignas o las heredadas de un padre normal; finalmente, se clasifican como VUS las que contienen uno o más genes con funciones mal definidas, contienen elementos reguladores o las que no encajan en las otras dos categorías20. Ha habido cierta preocupación entre los laboratorios clínicos por la posibilidad de malinterpretaciones del término de VUS que supongan un carácter más benigno de lo previsto.
Recientemente se ha sugerido que algunas CNV previamente consideradas benignas, pueden tener implicaciones patogénicas, por lo cual actualmente la mayoría de laboratorios incluyen en sus reportes las CNV benignas21. Muchos laboratorios utilizan un umbral de tamaño, desde 50 hasta 500 kilobases para reportar deleciones y desde 150 hasta 500 kilobases para reportar duplicaciones17. Esta aproximación reduce las VUS pero puede erróneamente excluir CNV clínicamente significativas de menor tamaño22.
COMPARACIÓN CON EL CARIOTIPO
El cariotipo es una técnica operador dependiente y el resultado tarda en promedio 14 días. En Colombia tiene un costo aproximado de 200 dólares, en Europa y Estados Unidos 300 euros y 500 dólares, respectivamente13 En 2010 se publicó un documento de consenso23 y un artículo de análisis económico24 sugiriendo al aCGH como primera prueba diagnóstica, reemplazando el cariotipo en pacientes con problemas neurológicos, autismo, déficit cognitivo o recién nacidos con anomalías congénitas de etiología desconocida. Análisis posteriores comparando aCGH, cariotipo, MLPA y QF-PCR, encontraron ventajas del aCGH sobre las otras pruebas, al examinar todo el genoma con mejor resolución para la detección de pérdidas o exceso de material genético25.
Aunque el resultado del aCGH está disponible en cuatro a siete días, el proceso es sistematizado y no es operador dependiente, este puede arrojar resultados no concluyentes como VUS, no detecta rearreglos balanceados, ni poliploidias (sí detectados por el cariotipo), no detecta mutaciones puntuales (tampoco detectadas en el cariotipo)26, y tiene como importante problema el costo, que en Colombia oscila entre 1500 y 3000 dólares, y en Europa y Estados Unidos es aproximadamente 500 euros y 1500 dólares respectivamente13.
ETIOLOGÍA DEL RETARDO MENTAL IDIOPÁTICO
Las alteraciones cromosómicas numéricas y estructurales, además de originar RM, se asocian a problemas del crecimiento, dismorfismo facial, malformaciones estructurales cardíacas, urogenitales y de las extremidades; al conjunto de estas características se le denomina fenotipo cromosómico. En particular, el RM se ha asociado a alteraciones en prácticamente todos los cromosomas. La citogenética estándar con 400-500 bandas, encuentra anomalías en el 40% de pacientes con RM grave y en el 10% de pacientes con retardo mental leve27. Un estudio canadiense encontró un 27% de diagnóstico positivo del aCGH sobre un 19% del cariotipo al usarlos para evaluar pacientes con RM28. En la mayoría de los trabajos, la trisomía 21 es la principal causa, seguida del síndrome X frágil29.
El RM idiopático es aquel al cual se le realiza historia clínica, pruebas genéticas y otros estudios sugeridos, y no se logra obtener una causa definitiva que lo explique. En la población mundial más del 50% de los casos de RM leve se clasifican como idiopáticos30. En Colombia no se conoce la epidemiología de esta entidad31. Una proporción importante de los afectados tienen anomalías cromosómicas submicroscópicas o defectos en genes únicos que juegan un papel en la etiología, predominando las aneuploidías cromosómicas, alteraciones estructurales con pequeñas deleciones terminales e intersticiales, duplicaciones terminales e intersticiales, los cromosomas marcadores extras, las traslocaciones desequilibradas y las traslocaciones recíprocas aparentemente equilibradas32-4.
Desde 1995, se reconoce que una causa significativa de RM idiopático la constituyen las alteraciones crípticas de regiones subteloméricas que producen ganancias o pérdidas y provocan desequilibrio de dosis génica35. La frecuencia de alteraciones subteloméricas en casos de RM idiopático es del 5 al 7%33,36,37; sin embargo, se ha encontrado que en pacientes con RM idiopático, las anomalías cromosómicas intersticiales son incluso más frecuentes que las subteloméricas38-40.
Estas alteraciones son difíciles de detectar en el cariotipo36, y se requieren técnicas moleculares de cribado genómico como el aCGH, que ha probado su validez para hallar desequilibrios submicroscópicos41-3. Aunque esta técnica tiene la desventaja de no detectar reorganizaciones equilibradas ni mosaicismos de bajo porcentaje, es en la actualidad la que, estando al alcance del laboratorio clínico, permite una valoración más extensa del genoma.
SÍNDROMES DE MICRODUPLICACIÓN O MICRODELECIÓN
Las consecuencias fenotípicas de las alteraciones cromosómicas no están obligatoriamente asociadas al tamaño. Se sabe que el efecto fenotípico de una anomalía cromosómica pequeña, depende más del locus y función de los genes implicados que del tamaño33. Así, un RM puede originarse por alteraciones cromosómicas grandes como las detectables por cariotipo (cinco a diez megabases) o pequeñas como las detectables por aCGH (cinco a diez kilobases)13.
Los síndromes de genes contiguos son causados por ganancia o pérdida de regiones cromosómicas submicroscópicas específicas. Dado que hoy se sabe que aunque varios genes estén involucrados, es posible que solamente uno sea sensible a la dosis y por tanto el causante del fenotipo, la denominación "síndromes de genes contiguos" ha sido reemplazada por MMS. Las técnicas moleculares, particularmente el aCGH, han permitido identificar un número creciente de MMS, principalmente en pacientes con RM6,27. En la Tabla 1 se muestran algunos MMS y sus características principales.
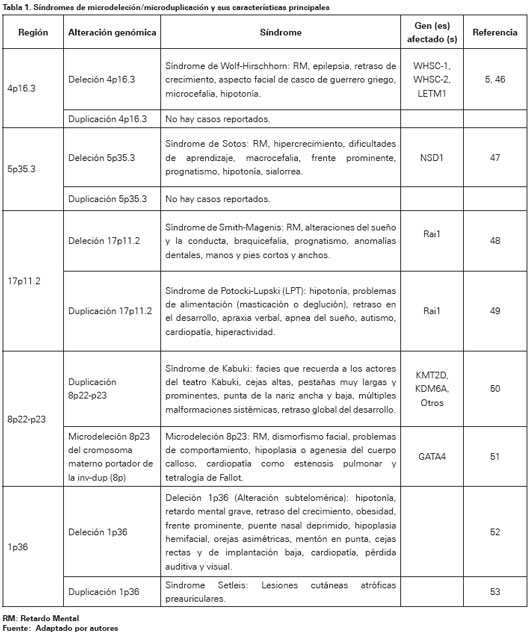
Teóricamente, para cada síndrome de microdeleción hay un síndrome de microduplicación recíproco. Sin embargo, se ha estimado una relación síndromes de microdeleción:síndromes de microduplicación de 2,5:1, y se ha reportado que solo para el 21% de los síndromes de microdeleción hay un síndrome recíproco de microduplicación44. En general, se puede afirmar que los MMS suelen incluir el RM dentro de sus fenotipos y que las microduplicaciones parecen producir fenotipos más leves o ninguno comparados con sus microdeleciones recíprocas45.
CAUSAS DE RECURRENCIA DE MMS
La base de la recurrencia de deleciones, duplicaciones, inserciones, inversiones y translocaciones, es la arquitectura del genoma humano19, específicamente la presencia de elementos repetitivos que permiten recombinaciones intercromátide, intercromosoma o intracromosoma ilegítimas durante la meiosis o la mitosis. Esta inestabilidad genómica no solamente causa MMS, sino que tiene gran impacto en la evolución del genoma y la flexibilidad para ganar nuevas funciones génicas o efectos directos por la dosis génica54,55. La ocurrencia de rearreglos genómicos recurrentes puede estar mediada por diferentes mecanismos, los cuales se muestran en la Tabla 2.
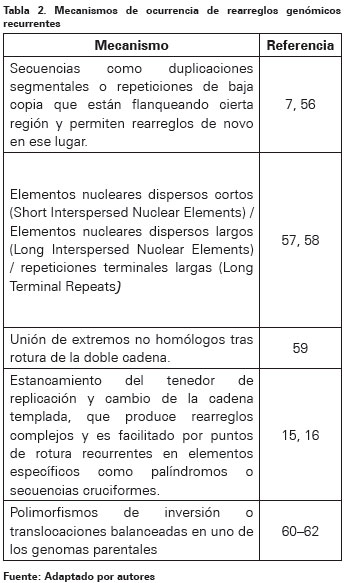
Dado que una causa común para microdeleciones y microduplicaciones recurrentes es la presencia en el genoma de innumerables repeticiones que pueden causar recombinación homóloga no alélica, el número total de MMS recíprocos puede ser mucho mayor al que ahora se conoce63.
HIBRIDACIÓN GENÓMICA COMPARATIVA CON MICROARRAYS VERSUS CARIOTIPO EN EL ANÁLISIS CROMOSÓMICO PRENATAL
El diagnóstico prenatal busca establecer el estado de salud del embrión o feto. Este puede ser no invasivo como la ecografía y las pruebas en sangre materna, o invasivo como la biopsia de vellosidad corial, la amniocentesis o la cordocentesis11; indicados estos últimos cuando la probabilidad de encontrar un diagnóstico anormal es mayor que el riesgo de complicación por el procedimiento64.
El cariotipo detecta prenatalmente alteraciones cromosómicas numéricas como trisomías, monosomías, poliploidias, y defectos estructurales como pérdidas o ganancias de material genético de cinco megabases, junto con translocaciones balanceadas. El cariotipo ha sido la prueba de oro en el análisis cromosómico prenatal en los últimos cuarenta años65. Sin embargo, recientemente se ha venido incrementando el uso de la aCGH y se han realizado comparaciones con el cariotipo, encontrando que las frecuencias de diagnóstico prenatal con cariotipo son de 2,55% a 4,16%, y para aCGH de 5,3%, a 15%. En general, en el diagnóstico prenatal el cariotipo tiene una sensibilidad de 73,36% y una especificidad de 99,86%, y los aCGH una sensibilidad de 98,21% con especificidad de 99,75%66.
CONCLUSIONES
Las técnicas moleculares, particularmente el aCGH, tienen mayor rendimiento diagnóstico que el cariotipo convencional en pacientes con RM idiopático debido a que permiten la detección de anomalías submicrocópicas, así como la identificación de un número creciente de MMS. Adicionalmente, el aCGH tiene mayor sensibilidad y una especificidad similar para diagnóstico prenatal de anomalías cromosómicas. Es probable que el aCGH se consolide como la prueba de primera línea para el análisis cromosómico en los próximos años. Incluso, ya se ha sugerido al aCGH como primera prueba diagnóstica, reemplazando el cariotipo en pacientes con problemas neurológicos, autismo, déficit cognitivo o recién nacidos con anomalías congénitas de etiología desconocida.
REFERENCIAS BIBLIOGRÁFICAS
1. Battaglia A, Carey J. Diagnostic evaluation of developmental delay/mental retardation: An overview. Am J Med Genet. 2003;117C(1):3-14. [ Links ]
2. Frey G, Temple V. Health promotion for Latin Americans with intellectual disabilities. Salud Publica Mex. 2008;50(2):167-77. [ Links ]
3. Salvador-Carulla L, Rodríguez-Blázquez C, Martorell A. Intellectual disability: an approach from the health sciences perspective. Salud Publica Mex. 2008;50(2):142-50. [ Links ]
4. Saldarriaga W, Ballesteros A. Diagnóstico prenatal de anomalías genéticas. Texto Neonatol. 2012:497-502. [ Links ]
5. Bergemann A, Cole F, Hirschhorn K. The etiology of Wolf- Hirschhorn syndrome. Trends Genet. 2005;21(3):188-95. [ Links ]
6. Weise A, Mrasek K, Kosyakova N, Mkrtchyan H GM, V. K. Fluorescence in situ hybridization (FISH)- application guide. In: Liehr T, editor. ISH probes derived from BACs, including microwave treatment for better FISH. In: Berlin Heidelberg, Germany: Springer-Verlag. Vol Chapter 4. ; 2009. [ Links ]
7. Stankiewicz P, Lupski J. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18(2):74-82. [ Links ]
8. Ji Y, Eichler E, Schwartz S, Nicholls RD. Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res. 2000;10(5):597-610. [ Links ]
9. Pujana MA, Nadal M, Guitart M, Armengol L, Gratacós M, Estivill X. Human chromosome 15q11-q14 regions of rearrangements contain clusters of LCR15 duplicons. Eur J Hum Genet. 2002;10(1):26-35. [ Links ]
10. Artigas-Pallarés J, Gabau-Vila E, Guitart-Feliubadaló M. El autismo sindrómico: I. Aspectos generales. Rev Neurol. 2005;40(Supl 1):S143-49. [ Links ]
11. Saldarriaga W, Ballesteros A. Diagnóstico Prenatal de Anomalías Genéticas. Distribuna. Bogota; 2012. [ Links ]
12. Leung T, Pooh R, Wang C, Lau T, Choy K. Classification of pathogenic or benign status of CNVs detected by microarray analysis. Expert Rev Mol Diagn. 2010;10(6):717-21. [ Links ]
13. Saldarriaga-Gil W. De la observación microscópica de los cromosomas en el cariotipo a los array-CGH en el diagnóstico prenatal. Rev Colomb Obstet Ginecol. 2013;64(3):327-32. [ Links ]
14. Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20(2):207-11. [ Links ]
15. Lee J, Carvalho C, Lupski J. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131(7):1235-47. [ Links ]
16. Lee C, Iafrate A, Brothman A. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39(7):48-54. [ Links ]
17. Tsuchiya K, Shaffer L, Aradhya S, Gastier J, Patel A, Rudd M, et al. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet Med. 2009;11(12):866-73. [ Links ]
18. Rodriguez-Revenga L, Mila M, Rosenberg C, Lamb A, Lee C. Structural variation in the human genome: The impact of copy number variants on clinical diagnosis. Genet Med. 2007;9(9):600-6. [ Links ]
19. Weise A, Mrasek K, Klein E, Mulatinho M, Llerena JC Jr, Hardekopf D, et al. Microdeletion and Microduplication Syndromes. J Histochem Cytochem. 2012;60(5):346-58. [ Links ]
20. McKusick-Nathans Institute of Genetic Medicine. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University (Baltimore, MD). http://omim.org/. Accessed August 15, 2011. [ Links ]
21. von der Lippe C, Rustad C, Heimdal K, Rødningen OK. 15q11.2 microdeletion - Seven new patients with delayed development and/or behavioural problems. Eur J Med Genet. 2011;54(3):357-60. [ Links ]
22. Kearney H, South S, Wolff D, Lamb A, Hamosh A, Rao KW. American College of Medical Genetics recommendations for the design and performance expectations for clinical genomic copy number microarrays intended for use in the postnatal setting for detection of constitutional abnormalities. Genet Med. 2011;13(7):676-9. [ Links ]
23. Miller D, Adam M, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749-64. [ Links ]
24. Regier D, Friedman J, Marra C. Value for money? Array genomic hybridization for diagnostic testing for genetic causes of intellectual disability. Am J Hum Genet. 2010;86(5):765-72. [ Links ]
25. Saldarriaga W, García-Perdomo HA, Arango-Pineda J, Fonseca J. Karyotype versus genomic hybridization for the prenatal diagnosis of chromosomal abnormalities: a metaanalysis. Am J Obstet Gynecol. 2015;212(3):330.e1-330.e10 [ Links ]
26. Mori M, Mansilla E, García F, Vallespín E, Palomares M, Martín R, et al. Diagnóstico prenatal y array-hibridación genómica comparada (CGH)(I). Gestaciones de elevado riesgo. Diagn Prenat. 2012;23(2):34-48. [ Links ]
27. Guitart-Feliubadaló M, Brunet-Vega A, Villatoro-Gómez S, Baena- Díez N, Gabau-Vila E. Causas cromosómicas que originan el retraso mental: alteraciones cromosómicas diagnosticables en el paciente. Rev Neurol. 2006;42(supl 1):S21-6. [ Links ]
28. Regier D, Friedman J, Marra C. Value for money? Array genomic hybridization for diagnostic testing for genetic causes of intellectual disability. Am J Hum Genet. 2010;86:765-72. [ Links ]
29. Van Karnebeek CDM, Jansweijer MCE, Leenders AGE, Offringa M, Hennekam RCM. Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness. Eur J Hum Genet. 2005;13(1):6-25. [ Links ]
30. Chelly J, Khelfaoui M, Francis F, Chérif B, Bienvenu T. Genetics and pathophysiology of mental retardation. Eur J Hum Genet. 2006;14(6):701-13. [ Links ]
31. Cabarcas Castro L. Etiologia del retardo mental en la poblacion pediatrica que asiste a consulta de neuropediatría en el hospital militar central e instituto de ortopedia infantil roosevelt bogota, colombia, 1 agosto 2009 - 31 de enero de 2011[TESIS]. Bogotá: Universidad Militar Nueva Granada;2011. [ Links ]
32. Battaglia A, Bianchini E CJ. Diagnostic yield of the comprehensive assessment of developmental delay/mental retardation in an institute of child neuropsychiatry. Am J Med Genet. 1999;82(1):60-6. [ Links ]
33. Bocian E, Helias-Rodzewicz Z, Suchenek K, Obersztyn E, Kutkowska-Kazmierczak A, Stankiewicz P, et al. Subtelomeric rearrangements: results from FISH studies in 84 families with idiopathic intellectual disability. Med Sci Monit. 2004;10(4):CR143-51. [ Links ]
34. Gribble SM, Prigmore E, Burford DC, Porter KM, Ng BL, Douglas EJ, et al. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Genet. 2005;42(1):8-16. [ Links ]
35. Flint J, Knight S. The use of telomere probes to investigate submicroscopic rearrangements associated with mental retardation. Curr Opin Genet Dev. 2003;13(3):310-6. [ Links ]
36. Koolen DA, Nillesen WM, Versteeg MH, Merkx GF, Knoers NV, Kets M, et al. Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet. 2004;41(12):892-9. [ Links ]
37. Rooms L, Reyniers E, Kooy R. Subtelomeric rearrangements in the mentally retarded: a comparison of detection methods. Hum Mutat. 2005;25(6):513-24. [ Links ]
38. Vissers LELM, Vries BBA De, Osoegawa K, Janssen IM, Feuth T, On Choy C, et al. Array-Based Comparative Genomic Hybridization for the Genomewide Detection of Submicroscopic Chromosomal Abnormalities. Am J Hum Genet. 2003;73(6):1261-70. [ Links ]
39. Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler HE, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/ mental retardation and dysmorphic features. J Med Genet. 2004;41(4):241-8. [ Links ]
40. Schoumans J, Ruivenkamp C, Holmberg E, Kyllerman M, Anderlid B-M, Nordenskjöld M. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH). J Med Genet. 2005;42(9):699-705. [ Links ]
41. Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J, et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet. 2001;29(3):263-4. [ Links ]
42. Kriek M, White S, Bouma M, Dauwerse H, Hansson K, Nijhuis J. Genomic imbalances in mental retardation. J Med Genet. 2004;41(4):249-55. [ Links ]
43. Bejjani BA, Theisen AP, Ballif BC SL. Array-based comparative genomic hybridization in clinical diagnosis. Expert Rev Mol Diagn. 2005;5(3):421-9. [ Links ]
44. Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Martyn L. Europe PMC Funders Group The rates of de novo meiotic deletions and duplications causing several genomic disorders in the male germline. Nat Genet. 2009;40(1):90-5. [ Links ]
45. Liehr T, Ewers E, Hamid AB, Kosyakova N, Voigt M, Weise A, et al. Small supernumerary marker chromosomes and uniparental disomy have a story to tell. J Histochem Cytochem. 2011;59(9):842-8. [ Links ]
46. Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, et al. Mapping the Wolf-Hirschhorn Syndrome Phenotype Outside the Currently Accepted WHS Critical Region and Defining a New Critical Region , WHSCR-2. Am J Hum Genet. 2003;72(3):590-7. [ Links ]
47. Tatton-Brown K, Douglas J, Coleman K, Baujat G, Chandler K, Clarke A, et al. Multiple mechanisms are implicated in the generation of 5q35 microdeletions in Sotos syndrome. J Med Genet. 2005;42(4):307-13. [ Links ]
48. Chen L, Mullegama S V, Alaimo JT, Elsea SH. Smith-Magenis syndrome and its circadian influence on development, behavior, and obesity - own experience. Dev period Med. 19(2):149-56. [ Links ]
49. Magoulas PL, Liu P, Gelowani V, Soler-Alfonso C, Kivuva EC, Lupski JR, et al. Inherited dup(17)(p11.2p11.2): expanding the phenotype of the Potocki-Lupski syndrome. Am J Med Genet A. 2014;164A(2):500-4. [ Links ]
50. Cheon C-K, Ko JM. Kabuki syndrome: clinical and molecular characteristics. Korean J Pediatr. 2015;58(9):317-24. [ Links ]
51. Ballarati L, Cereda A, Caselli R, Selicorni A, Recalcati MP, Maitz S, et al. Genotype-phenotype correlations in a new case of 8p23.1 deletion and review of the literature. Eur J Med Genet. 54(1):55-9. [ Links ]
52. Jordan VK, Zaveri HP, Scott DA. 1p36 deletion syndrome: an update. Appl Clin Genet. 2015;8:189-200. [ Links ]
53. Lee BH, Kasparis C, Chen B, Mei H, Edelmann L, Moss C, et al. Setleis syndrome due to inheritance of the 1p36.22p36.21 duplication: evidence for lack of penetrance. J Hum Genet. 2015;60(11):717-22. [ Links ]
54. Gazave E, Darré F, Morcillo-Suarez C, Petit-Marty N, Carreño A, Marigota UM, et al. Copy number variation analysis in the great apes reveals species-specific patterns of structural variation. Genome Res. 2011;21(10):1626-39. [ Links ]
55. Marques-Bonet T, Eichler E. The Evolution of Human Segmental Duplications and the Core Duplicon Hypothesis. Cold. 2009;74:355-62. [ Links ]
56. Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7(7):552-64. [ Links ]
57. Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, et al. Europe PMC Funders Group Paired-End Mapping Reveals Extensive Structural Variation in the Human Genome. Science. 2009;318(5849):420-6. [ Links ]
58. Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453(7191):56-64. [ Links ]
59. Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283(1):1-5. [ Links ]
60. Gimelli G. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12(8):849-58. [ Links ]
61. Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38(9):999-1001. [ Links ]
62. Shaffer LG. Diagnosis of microdeletion syndromes by fluorescence in situ hybridization (FISH). Curr Protoc Hum Genet. 2001; Capitulo 8:Unidad 8.10. [ Links ]
63. Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1(1):4. [ Links ]
64. Gratacos E, Delgado J. Procedimientos invasivos ecoguiados en medicina fetal. Med fetal. 2007:129-135. [ Links ]
65. Brady P, Devriendt K, Deprest J, Vermeesch J. Array-Based Approaches in Prenatal Diagnosis. Methods Mol Biol. 2012;838:151-71. [ Links ]
66. Armengol L, Nevado J, Serra-Juhé C, Plaja A, Mediano C, García-Santiago FA, et al. Clinical utility of chromosomal microarray analysis in invasive prenatal diagnosis. Hum Genet. 2012;131(3):513-23. [ Links ]

