











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La coroideremia es una enfermedad conocida hace más de 140 años, que en su descripción original no fue considerada una patología sistémica, pero si un trastorno ocular aislado1. Fue descrita por primera vez en 1872 por Mauthner, y en 1948 se descubrió su patrón ligado al cromosoma X2. La incidencia de este trastorno ocular primario varía entre 1:50 000 a 1:100 000 personas3. La prevalencia reportada es de 1:50 000 personas de descendencia europea3,4. No se conoce la prevalencia e incidencia de dicha entidad en Colombia, sin embargo, existen múltiples casos reportados en la literatura mundial donde describen sus hallazgos característicos.
El término coroideremia se refiere literalmente a la ausencia (eremia) de la coroides5, capa del globo ocular que hace parte a su vez del tracto uveal (conformado por el iris, cuerpo ciliar y coroides). La coroideremia es una degeneración progresiva coriocapilar del Epitelio Pigmentario Retiniano (EPR) y de los fotorreceptores de la retina, causada por una mutación en el gen que codifica para la proteína REP- 1, localizada en el cromosoma Xq216,7. Posee herencia ligada al X, afectando a los hombres más gravemente que a las mujeres portadoras, quienes pueden mostrar cambios pigmentarios característicos al fondo de ojo con degeneración coriorretiniana irregular8,9.
La coroideremia o distrofia tapetocoroidea progresiva, es una causa no clásica o no común de discapacidad visual (término que agrupa las deficiencias visuales permanentes tipo baja visión y ceguera), por tener una baja incidencia. La fisiopatología actual de la coroideremia se explica desde las mutaciones en el gen CHM que codifica para la proteína REP-1, localizada en el cromosoma Xq21. El gen CHM tiene 653aa, se expresa en bastones y EPR, pero no en conos, sugiriendo ser los bastones el sitio principal de afectación en la coroideremia10,11. Además, la REP-1 se expresa en todas las células tisulares del cuerpo humano, incluyendo la retina y los fotorreceptores, pero su isoforma REP-2 no se expresa en el ojo, y es por esto por lo que a pesar de que la función de REP-1 pueda ser compensada por REP-2, en el globo ocular no ocurre de este modo5,12. No está claro si la pérdida de fotorreceptores es secundaria a la degeneración del EPR o si puede generarse por sí sola, debido a defectos en el gen CHM, que también se expresa en estas células, sin embargo, la teoría más fuerte es que su origen se encuentra en el EPR13. Otros autores aducen que la degeneración del EPR o atrofia coroidea precede al daño de los fotorreceptores14.
La proteína REP-1 es necesaria para mediar la interacción entre Rab, molécula que se une a vesículas y compartimientos de transporte celular (mediante un grupo geranil-geranilo, y la enzima Rab geranilgeraniltransferasa). Una vez funcional (gracias a una geranilación), las proteínas Rab son menos solubles en el citoplasma celular y se activan por la unión a guanosín trifosfato, adquiriendo así la capacidad de unirse a la membrana celular, asegurando un efectivo transporte de vesículas15 que permite la viabilidad celular. Adicional a su rol en la regulación de los procesos de tráfico celular de vesículas, las proteínas Rab tienen una función de señalización a otros organelos celulares16: algunas Rab están implicadas en el transporte vesicular de la rodopsina desde el aparato de Golgi hasta el cilio de conexión en las células fotorreceptoras17. La falta de REP1 conduce a una ausencia de Rab27a funcional, específicamente en el epitelio pigmentario de la retina. La Rab27 es una importante proteína ocular, necesaria para la exocitosis de gránulos líticos en linfocitos citotóxicos, la regulación de la distribución periférica de melanosomas y la producción de los discos del segmento externo de los bastones (que contienen opsina). Esta proteína participa en la recuperación de dichos discos a través de la actividad fagocítica de las células del EPR y del transporte y eliminación de los productos finales de la fagocitosis (gránulos de lipofuscina)18. Al igual que los melanocitos, las células del EPR tienen gránulos que contienen melanina, y las Rab27 pueden reclutar un motor molecular basado en actina para orientar los gránulos que contienen melanina en el citoplasma, un evento que es esencial para la función y dinámica apropiada del EPR18.
La Rab27a se acumula en su forma no prenilada en el citosol en las células de los pacientes con coroideremia, y la pérdida de la función REP-1 reducirá principalmente la eficacia de prenilación de Rab27a y otros Rabs de baja afinidad (Rab27B, Rab38 y Rab42). Es así como la degeneración del EPR y sus capas adyacentes puede ser causado por un transporte melanosómico deficiente, lo que genera una falta de protección contra la exposición a la luz nociva16,17.
En el EPR anormal hay células de grosor irregular que contienen un número variable de gránulos de melanina y lipofuscina, incluso en regiones donde el EPR está bien conservado. Algunos autores han demostrado que en la coroideremia se evidencia la reducción de la acidificación lisosómica y retrasos en la degradación de fagosomas de las células del EPR. Lo anterior desencadena una acumulación de material similar a la lipofuscina, aspecto que provoca la degeneración de las células del EPR y como consecuencia, pérdida progresiva de la Agudeza Visual (AV) hasta ceguera19.
La retina disminuye su grosor debido al adelgazamiento de los segmentos externos de los fotorreceptores y a la pérdida de núcleos dentro de la capa nuclear externa e interna. Diferentes autores plantean la teoría de que existe cierto grado de inflamación dentro de la retina en pacientes con coroideremia. Un informe reciente ha proporcionado pruebas de un leve infiltrado de linfocitos T dentro de la coroides20.
Las mutaciones del gen CHM pueden dar como resultado una deleción completa o un producto génico no funcional, causando truncamiento, perdida de dominio funcional o ausencia de la proteína6,9,19,21. Hasta el 15% de las mutaciones en el gen CHM son deleciones totales o parciales, donde incluso las deleciones de genes contiguos en el cromosoma X, que incluyen la supresión de Xq21, puedan causar casos de coroideremia.
En los hombres la coroideremia se caracteriza por producir inicialmente nictalopía (ceguera o disminución de la visión en la noche o en ambientes de baja iluminación) en la infancia, y pérdida progresiva de la visión periférica más AV con el paso de los años. Los campos visuales en última instancia se reducen arduamente a un área central de 5° a 10° debido al compromiso foveal. La AV disminuye a categorías de deficiencia visual moderada y grave (AV entre 20/60 y 20/400), progresando en fase tardía a ceguera (AV menor a 20/400 hasta no percepción de luz). Sin embargo, los pacientes pueden mantener una función visual central relativamente buena hasta los 50 a 70 años2,9, 20.
Durante la evolución de esta entidad, hay cambios pigmentarios con atrofia coroidea focal alrededor de la zona ecuatorial, patrón que se conoce como “manchas de pigmento sal y pimienta”. Eventualmente la degeneración progresa al área central y mácula, que se mantiene intacta por varias décadas. Las mujeres portadoras generalmente son asintomáticas con campos visuales conservados, pero en el fondo de ojo se puede observar una imagen en “parche” con grupos de pigmento en el fondo de la periferia media y del polo posterior. El examen de fondo de ojo es fundamental en este tipo de pacientes puesto que la observación de amplias áreas de atrofia coroidea, vasos coroideos y esclerótica, más la preservación de la región macular, son característicos8,9 .
El diagnóstico de la coroideremia se basa en los signos y síntomas clínicos, el aspecto característico del fondo de ojo, el patrón de degeneración de los conos y los bastones en el electrorretinograma y un antecedente familiar consistente con una herencia ligada al cromosoma X. Es posible realizar el diagnóstico genético analizando el ARNm y ADN del gen CHM. Adicionalmente, se pueden hacer otros estudios como la angiografía con fluoresceína intravenosa, tomografía ocular coherente y electrorretinograma. En la angiografía con fluoresceína intravenosa, las áreas de atrofia coroidea se caracterizan por un fondo hipofluorescente y líneas hiperfluorescentes que representan los vasos coroideos profundos conservados, y la tomografía ocular coherente logra evidenciar un engrosamiento alrededor de la región parafoveal y posteriormente en la fóvea, un adelgazamiento periférico excéntrico y finalmente un adelgazamiento central y periférico generalizado8,9. Los diagnósticos diferenciales principales son la retinosis pigmentaria, síndrome de Usher tipo 1 y la atrofia generalizada de la coroides y de la retina.
El manejo de esta entidad incluye exámenes oftalmológicos periódicos, el uso de gafas de sol que bloqueen la radiación UV, y la remisión y seguimiento de los pacientes por parte de especialistas en baja visión2,9,20. La adición de suplementos con vitaminas antioxidantes (incluyendo vitamina A), omega-3 y ácido docosahexanoico pueden ser benéficos22. Actualmente no hay tratamiento disponible, pero se está desarrollando un ensayo clínico para tener disponibilidad de la terapia génica, que tiene como objetivo retardar la degeneración ocasionada por la enfermedad23. La terapia consiste en introducir adenovirus que contienen la región codificadora de CHM capaz de restaurar los niveles de proteína in vitro y la actividad REP-1 en linfocitos y fibroblastos deficientes en la enfermedad23,24.
En Latinoamérica se han reportado dos familias mexicanas a quienes se les realizó un estudio del gen CHM. Dicho estudio encontró dos mutaciones: mutación R270X (transición de C a T en el nucleótido 808 en el exón 6) y mutación S468X (transversión C a G en la posición de nucleótidos 1403 en el exón 11). La segunda mutación no había sido descrita anteriormente en la literatura25.
A pesar de que la enfermedad está bien estudiada en el campo de la genética oftalmológica, el objetivo del presente artículo consiste en reportar las características clínicas, el proceso de deterioro visual y adquisición de discapacidad visual de un paciente con coroideremia familiar residente en Medellín, Antioquia-Colombia. Se dio aprobación y diligenciamiento de consentimiento informado por parte del paciente.
Presentación del caso
Hombre de 54 años soltero, natural de la ciudad de Medellín/Colombia, sin hijos, con escolaridad primaria y quien ha laborado en oficios varios. Desde su infancia presenta problemas de visión nocturna tipo sensación de deslumbramiento con luces, que han afectado la realización de sus oficios. El paciente comenta que además del deslumbramiento con luces, tiene que girar la cabeza para compensar la visión de su Ojo Derecho (OD). No reporta la ocurrencia de accidentes automovilísticos, sin embargo, refiere no haber manejado de noche. Refiere que cuando cumplió 28 años el deterioro visual fue progresivo, donde la sintomatología comprometió primero al OD y a la edad de 34 años afectó su Ojo Izquierdo (OI). Luego de indagación diagnóstica, y a partir de una angiografía (Ver Figura 1), fue diagnosticado con coroideremia a la edad de 36 años.
A partir del diagnóstico de coroideremia se hizo hincapié en sus antecedentes familiares, donde su madre (natural de Marinilla, oriente de Antioquia) usa lentes desde los 17 años, padre (natural de Medellín) sin aparente alteración visual. Sin embargo, el paciente mencionó que un hermano y un primo materno también presentaban deterioro visual; el hermano presenta diagnóstico de coroideremia (Ver Figura 2).
Cuando el paciente cumplió 45 años fue necesario realizar una nueva angiografía y un electrorretinograma, al presentar nueva sintomatología visual, que consistió en: disminución de AV en OD movimiento de manos a 10 cm y OI 20/50 (estenopeico sin mejoría). El médico tratante sugirió presencia de membrana neovascular en OD. El resultado del electrorretinograma fue sin respuesta en los tres componentes electrofisiológicos de la visión. (Ver Figura 3)
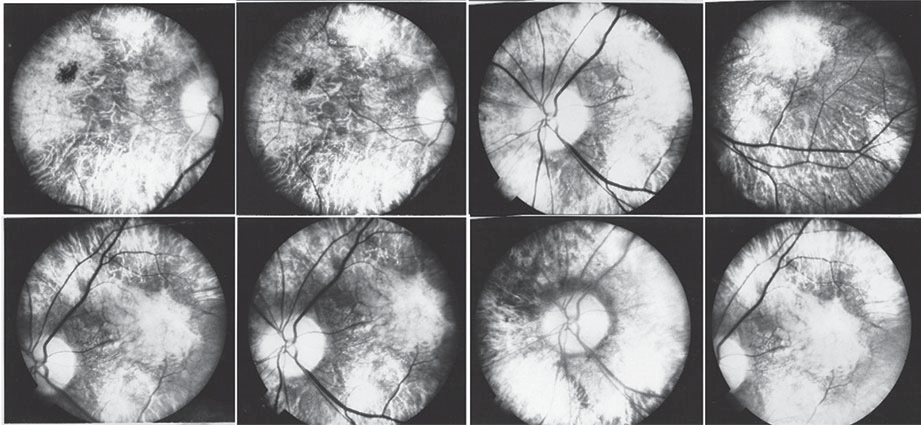
Fuente: autores.
Figura 1. Angiografía inicial. Atrofia del epitelio pigmentario de la retina en OD (primera fila de imágenes) que permite la visión de los vasos coroideos y la esclera, atrofia óptica y arterias delgadas, y daño del área macular. En OI (segunda fila de imágenes) hubo hallazgos similares, menos atrofia marcada del EPR y no atrofia óptica. En ambos ojos se aprecia atrofia del EPR, y en el OD presenta hipertrotrofia del EPR superior y temporal a la mácula, con adelgazamiento retiniano que permite apreciar la vasculatura coroidea. En el área macular se observa cambios atróficos y migración pigmentaria generalizada. Además, se observa adelgazamiento del calibre de los vasos retinianos. OD presenta cambios atróficos del nervio óptico y OI nervio óptico sin atrofia.
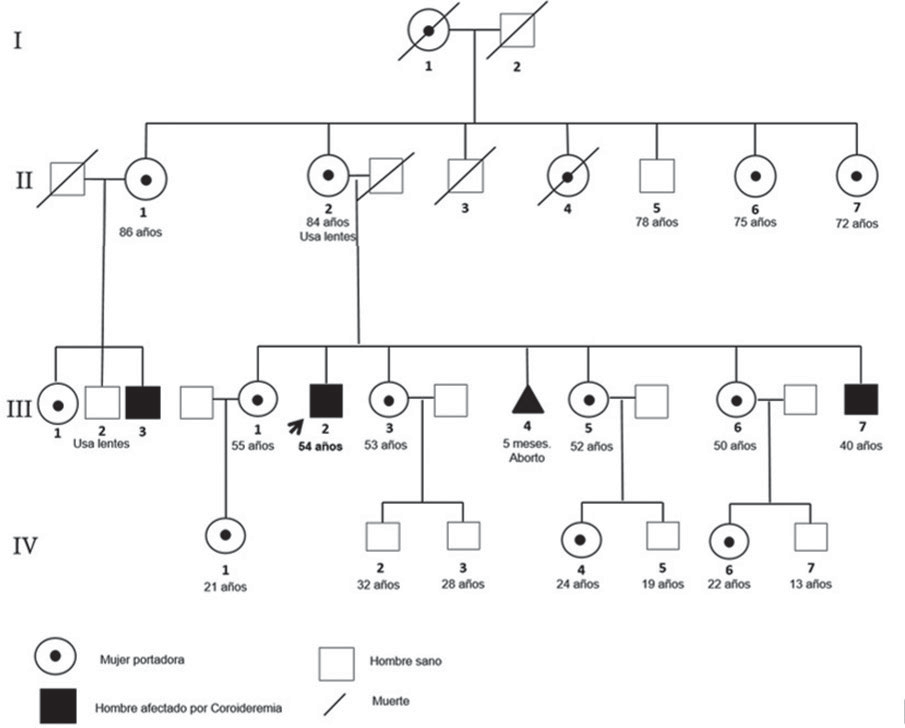
Fuente: autores.
Figura 2. Familiograma. Caso índice (flecha). Hermano menor soltero y sin hijos, tiene diagnóstico de coroideremia (síntomas desde los 13 años y actualmente no labora por su condición de salud). El paciente y su hermano poseen 7 sobrinos (por parte de sus cuatro hermanas), cuatro de ellos son hombres los cuales no han presentado síntomas compatibles con nictalopía. Adicionalmente, se informa de un primo materno con sospecha diagnóstica de coroideremia.
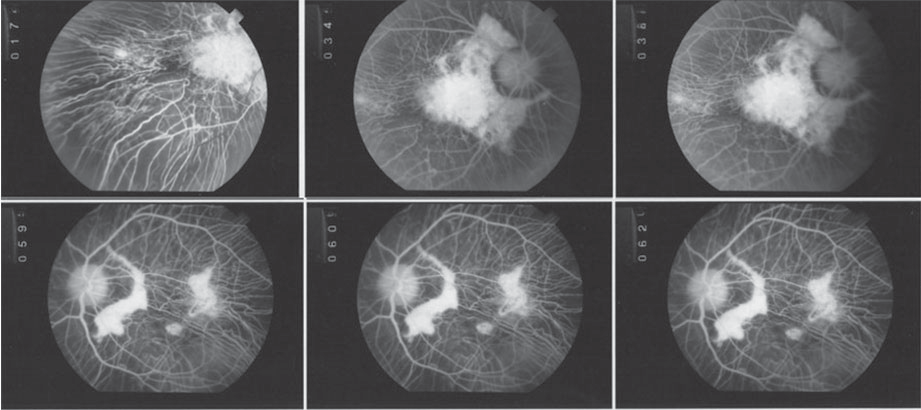
Fuente: autores.
Figura 3 Angiografía de seguimiento. Tanto en OD (primera fila de imágenes) como en OI (segunda fila de imágenes) se aprecia marcada progresión de la atrofia, respecto a la angiografía descrita previamente. Ambos ojos presentan atrofia coroidea grave, que permite visualizar las estructuras esclerales; en el OD compromete mácula severamente en el haz papilomacular y en el OI la misma lesión, más pequeña, arqueada, concéntrica a la papila. Además, se observa adelgazamiento del calibre de los vasos retinianos.
A la edad de 47 años le fue practicado una tomografía óptica coherente, la cual reportó en OD pérdida del contorno foveal, adelgazamiento retinal entre los 6 mm centrales, EPR coriocapilar engrosado a nivel subfoveal con un sobre levantamiento y lesión hiporreflectiva que pudiese corresponder bien sea a una masa coroidea o a membrana neovascular, y a su alrededor áreas de aumento de reflectividad en capas profundas que corresponden a zonas de atrofia del EPR. El anterior hallazgo, determinado mediante una prueba diagnóstica, fue corroborado por retinólogo al describir un fondo de ojo OD con palidez del disco óptico, atenuación de los vasos, espículas periféricas, atrofia difusa del EPR y membrana neovascular, y en OI palidez del disco, atenuación de los vasos, espículas escasas periféricas y atrofia generalizada del EPR. En otras palabras, el paciente entre los 47 a 51 años ya presentaba una marcada atrofia del EPR, y gran compromiso de la función visual.
Finalmente, el paciente a la edad de 54 años fue remitido a evaluación y manejo por optometría en baja visión y rehabilitación visual; cuenta con una AV en OD solo de movimiento de manos a 40 cm, y en OI una AV con corrección lejana de 20/64 y con corrección cercana de 20/100 (en la categoría de deficiencia visual permanente tipo baja visión). El manejo terapéutico se ha centrado en control oftalmológico para ver el progreso de la enfermedad, y utilización tanto de ayudas ópticas (tipo lupas y telescopios) como no ópticas (bastón de orientación y lentes con filtros). El paciente es autónomo en actividades básicas, evita deambular en lugares desconocidos, camina sin apoyo en lugares conocidos (por voluntad propia no utiliza bastón de orientación/movilidad), y posee dificultad en lectura de medios escritos.
Discusión
La coroideremia es una enfermedad ligada al sexo, que se transmite genéticamente a través del cromosoma X. Las mujeres tienen dos cromosomas X, y pueden llevar el gen afectado en uno de ellos, sin embargo tienen una versión sana de este gen y por lo tanto se comportan como portadoras26,27. Esto se ve reflejado en el paciente y familia aquí reportadas, donde hay mujeres portadoras, dos hombres afectados y un primo materno posiblemente afectado.
Los pacientes con este trastorno ocular que afecta el funcionamiento (siendo así una potencial causa de discapacidad visual), presentan síntomas tempranos en la primera y segunda década de la vida como lo es la nictalopía, seguida de una reducción de la visión periférica (5° a 10°)1. Este patrón clínico se evidenció en el paciente índice como en su hermano. A medida que avanza la degeneración del EPR y retina, se experimenta una marcada disminución de la AV hasta llegar a un estado de ceguera parcial y luego total (desde AV menores a 20/400 que no mejoran con corrección óptica hasta no percepción de luz) en la tercera a cuarta década de la vida. El paciente reportado posee un déficit visual permanente moderado en su mejor ojo (OI), cumpliendo criterios de discapacidad visual tipo baja visión, a pesar de que su OD se encuentra en rango de ceguera. Aunque algunos pacientes pueden tener defectos en la visión cromática, el paciente índice como su hermano no refieren esta dificultad, y en el seguimiento médico no fueron reportadas alteraciones de la visión de color. En México se han reportado dos familias25 en las cuales la nictalopía fue el síntoma inicial en los varones afectados con edades de inicio temprano (<20 años). La AV en los sujetos afectados varió de 20/20 a percepción de luz sin discriminación de color. Todas las mujeres portadoras a excepción de una de ambas familias mostraron AV normal25. Los hallazgos clínicos al fondo de ojo, por angiografía y tomografía óptica coherente del paciente aquí reportado, reconstruyen el deterioro progresivo de coroides y retina.
Algunos casos notificados incluyen pacientes con comorbilidades tipo retraso mental y motor, sordera neurosensorial, hendidura labial y palatina, y otros fenotipos clínicos (sin datos concisos que indiquen la frecuencia en pacientes con coroideremia). Un estudio de 2011 informó sobre un paciente que presentó coroideremia más displasia del sistema auricular, conducto arterioso permeable e hipoplasia del esmalte. Otro estudio informó coroideremia asociado con atrofia muscular espinal distal ligada al cromosoma X y síndrome de retraso mental con sordera Martin-Probst17. El paciente reportado en el presente artículo no presentaba deficiencias de orden mental, físico, cardiovascular ni sensorial auditivo al examen físico, ni en los reportes de historia clínica.
Todas las mutaciones hasta ahora conocidas y reportadas en coroideremia, conllevan a degeneración progresiva de los coriocapilares, EPR y fotorreceptores6,18. Las principales causas de ceguera en el mundo desarrollado son las enfermedades crónicas hereditarias de la retina, como la degeneración macular relacionada con la edad y la retinitis pigmentosa (RP)24; sin embargo, se debe tener presente que existen otras etiologías de menor frecuencia, como la coroideremia, que pertenece a las enfermedades hereditarias de la retina, la cual desde una óptica clínica y asistencial debe ser considerada.
El diagnóstico de coroideremia usualmente es clínico, basado en el examen de fondo de ojo, paraclínicos y en la historia familiar consistente con herencia ligada al cromosoma X. Esta fue la metodología aplicada en el actual reporte de caso, donde no se realizaron exámenes moleculares. En los pacientes afectados, el fondo de ojo puede revelar rasgos característicos de alteración del EPR y atrofia coriorretiniana, como quedó evidenciado en la descripción de las dos angiografías realizadas en el paciente. La manifestación típica de la coroideremia es la atrofia coroidea con festoneado y preservación de la mácula. Sin embargo, estos tipos de cambios típicos del fondo de ojo para la coroideremia pueden no haberse desarrollado todavía en la primera visita al oftalmólogo28.
Para el manejo de la coroideremia se recomiendan diferentes estrategias como el uso de lentes protectores de luz UV, revisión y seguimiento por especialistas en baja visión, suplementos multivitamínicos y terapia génica. En la literatura se ha reportado un ensayo clínico con seis hombres, donde se utilizó vectores virales adeno-asociado, REP1, encontrando una mejoría relativa en AV en dos de los seis pacientes, donde el incremento en la sensibilidad de la retina se correlacionó con la dosis del vector administrada4. En caso de presencia de agujero macular, el cual aparece por el desprendimiento de la retina, se puede realizar intervención quirúrgica donde se busca cerrar el orificio de forma anatómica para restaurar la integridad del espacio sub retiniano en la región de la mácula29.
Es importante tener en cuenta los diagnósticos diferenciales de coroideremia. Uno de ellos es la RP, en la cual hay anormalidades de conos, bastones y el EPR, llevando así a una pérdida progresiva visual, nictalopía y constricción del campo visual, siendo clínicamente similar a lo que ocurre en pacientes con coroideremia. En los últimos estadios de coroideremia, cuando hay pérdida de la coroides y retina, el examen del fondo de ojo puede ser confundido con un estado final de RP; sin embargo, el grado de la migración del pigmento en la retina que tipifica la RP no es visto en pacientes con coroideremia. La RP puede ser heredada de forma autosómica dominante, autosómica recesiva o ligada al cromosoma X. El patrón de autofluorescencia de imágenes retinianas en portadoras de RP ligada al X difieren en comparación a las portadoras de la coroideremia22,28.
Conclusiones
La coroideremia familiar es una enfermedad retiniana con herencia ligada al X que cursa con degeneración progresiva coriocapilar, del EPR y fotorreceptores, causada por una mutación en el gen que codifica para la proteína REP-1. Al ser una enfermedad ligada al X, explica el estatus de portador en las mujeres y mayor gravedad de signos y síntomas en los hombres.
El diagnóstico usualmente es clínico, basado en el examen de fondo de ojo (atrofia coriorretiniana, atrofia coroidea con festoneado y preservación macular), paraclínicos oftalmológicos, en la historia familiar consistente con herencia ligada al cromosoma X, y al tener presente siempre el diagnóstico diferencial más importante que es la RP y la degeneración macular relacionada con la edad.
Clínicamente es una causa potencial de discapacidad visual, que lleva paulatinamente a la persona a deficiencias visuales permanentes, primero en rango baja visión, y posteriormente con los años a deficiencias visuales completas tipo ceguera. De manera que en el manejo integral, es de vital importancia la necesidad de un diagnóstico precoz y el establecimiento de un equipo multidisciplinario para su abordaje (que incluya servicios de baja visión y rehabilitación).

