











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El Pénfigo Familiar Benigno (PFB) o la Enfermedad de Hailey-Hailey (EHH), es un tipo de dermatosis autosómica dominante descrita por primera vez en 1939 por los hermanos, Hugh y William Hailey, a los que se debe ahora su nombre. Tiene una prevalencia de alrededor de 1 en 50 000 casos, se ha reportado antecedentes familiares hasta en 60% de los pacientes siendo el 40% restante casos esporádicos1-2. Es causada por mutaciones en el gen ATPasa, transportador de Ca2+ tipo 2C, miembro 1 (ATP2C1)2-3-4, ubicado en el cromosoma 3q22.1, que codifica la ruta secretora humana Ca2+/ Mn2+ ATPasa proteína 1 (hSPCA1), y juega un papel importante en el control de la concentración de Ca2+ en el citoplasma y el aparato de Golgi de los queratinocitos humanos., lo que ocasiona principalmente un defecto en la adhesión. Los queratinocitos derivados del PFB presentan un aumento del estrés oxidativo, que se asocia con una proliferación y diferenciación deterioradas6. Factores precipitantes pueden originarse por estímulos provenientes del exterior (que impulsen la separación de los queratinocitos), dentro de ellos se puede encontrar: el calor, la fricción con objetos, luz solar, luz ultravioleta, radiación, entre otros7-8.
El PFB clínicamente se presenta con vesículas flácidas y ampollas recurrentes que determinan la formación de placas. En el centro de la lesión se forman pequeñas vesículas que se rompen dejando una superficie exudativa y erosiva. En los pliegues es común la presencia de maceraciones, con formas hipertróficas y vegetantes. Además, el signo de Nikolsky suele ser positivo en las áreas afectadas. El curso clínico suele ser por brotes que duran meses y curan sin dejar cicatrices, dejando en algunos casos hiperpigmentación residual9-10. En gran número de pacientes las lesiones suelen estar presente en la totalidad de la piel, la topografía habitual es en los grandes pliegues (axilas, ingles, región inframamaria, antecubital y poplítea) y en el cuello, sitio donde suelen comenzar las lesiones10. Presentaciones atípicas se han reportado en los hombros, la espalda, la cara y región pectínea. Los síntomas acompañantes más frecuentes son olor fétido, prurito y dolor, los cuales pueden afectar las actividades diarias, laborales y sociales. Dado que las manifestaciones clínicas son amplias e inespecíficas, es común un retraso en el diagnóstico, siendo erróneamente diagnósticado como dermatitis seborreica cuando se localiza en piel cabelluda o candidiasis en región perianal. Como diagnósticos diferenciales deben tenerse en cuenta un impétigo, pénfigo vulgar, la enfermedad de Darier-White, la enfermedad de Grover, dermatosis acantolítica transitoria o una dermatitis de contacto alérgica por los medicamentos tópicos utilizados11-12.
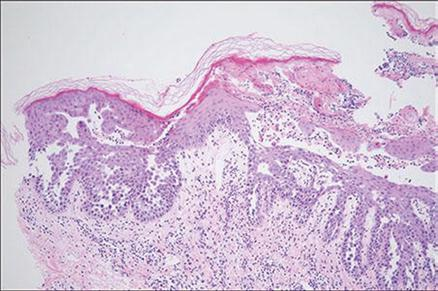
El estudio histológico es el examen de elección.. En él se puede identificar la formación de hendiduras o ampollas intraepidérmicas secundarias a acantolisis suprabasal, la cual generalmente es extensa y afecta gran parte del estrato espinoso. Se alteran los puentes intercelulares, dando una figura que asemeja a una “pared de ladrillos dilapidada”10. La acantolisis respeta el epitelio de las estructuras anexiales. Como contenido de las ampollas pueden observarse células acantolíticas aisladas o en grupos y es común observar papilas dérmicas elongadas y cubiertas por una o múltiples capas de queratinocitos semejando vellosidades11-12.
En la actualidad no existe tratamiento específico ni totalmente efectivo contra la enfermedad. Tampoco existen medidas profilácticas efectivas, solo aquellas encaminadas a proteger la piel de la maceración, el calor y demás factores desencadenantes como la fricción. El uso de antibióticos a bajas dosis se emplea para evitar sobreinfección bacteriana. La mayoría de los pacientes responden al tratamiento tópico con corticoides en monoterapia o combinado con antibióticos (tópicos o sistémicos) tipo aminoglucósidos, penicilinas, tetraciclinas y/o antifúngicos. Siendo estos útiles en el manejo de enfermedad aguda. Los inhibidores tópicos de calcineurina, como Tacrolimus 0.1%, se han usado con buenos resultados como terapia de mantenimiento. Otras opciones tópicas incluyen 5 F-U y Análogos de vitamina D. En caso de enfermedad extensa, recidivante y refractaria al tratamiento inicial, se puede plantear el empleo de retinoides orales como acitretina, isotretinoína y etretinato, aunque se desconoce el mecanismo. Otras opciones incluyen corticosteroides orales, ciclosporina, Metotrexato, Dapsona o terapia biológica con los Inhibidores del factor de necrosis tumoral α, como etanercept. Otras opciones de tratamiento en caso refractarios, incluyen modalidades quirúrgicas, como la escisión seguida de autoinjerto de piel, criocirugía, dermoabrasión. Todas ellas sin suficienciente evidencia12-13-14.
La evolución de la enfermedad suele estar dado por su cronicidad y periodos intercríticos dados por brotes que pueden durar meses. Muchos pacientes suelen mejorar con la edad. Aunque se considera una enfermedad con baja mortalidad su carga mórbida es muy alta y está dada por síntomas como dolor, ardor, prurito y olor desagradable. El dolor es el síntoma más molesto y el prurito el más frecuente12-13. Aunque no se altera el estado general de los pacientes, muchos refieren que su enfermedad interfiere con las actividades diarias, afectando su calidad de vida10-11-12-13.
El interés de presentar este artículo tiene como objetivos principales primero, describir esta entidad ante el escaso número de reportes de esta dermatosis en nuestro País. Segundo, aportar a la literatura el éxito terapéutico obtenido con corticoides sistémicos en PFB en un paciente de sexo masculino de 46 años de edad, que presentaba recurrencias desde hace 2 años, con lesiones polimorfas localizadas en zonas infrecuentes y con tratamiento favorable que le permitió al paciente mejorar significativamente su calidad de vida sin evidencia de efectos adversos por uso prolongado del tratamiento.
Caso clínico
Se reporta el caso de un hombre de 46 años de edad, procedente de área rural (Lebrija-Santander), de oficio agricultor, obeso, sin antecedentes familiares de Pénfigo familiar benigno, que acude al servicio de dermatología de forma ambulatoria por lesiones en piel de nuca y región deltoidea crónicas y recidivantes. Manifestó que el cuadro inicio hace 2 años, como vesículas y ampollas en pliegues que lo motivaron a consultar al hospital regional, donde diagnosticaron dermatitis de contacto, dada su condición de agricultor, siendo manejado con antihistamínico y corticoide tópico sin mejoría alguna de las lesiones, con múltiples remisiones al servicio de dermatología para valoraciones, las cuales no eran autorizadas por su EPS. Las lesiones fueron progresando a placas y exulceraciones asociadas a prurito intenso, que el paciente asociaba con las jornadas laborales y el clima seco. En los últimos 5 meses presentó aumento del número de lesiones, comprometiendo alrededor del 30 % de su superficie corporal, con predominio de extremidades superiores (hombros, espalda, tórax anterior) y pliegues, por lo que decide consultar al dermatólogo particular. En el examen físico se evidencia obesidad grado I (IMC 30), en piel presentaba lesiones tipo placa, de tamaño variable, desde 4 mm hasta 12mm en su mayor diámetro, de tonalidad rosada, bordes irregulares, con escama fina, áreas de exulceración y costras hemáticas. También había lesiones secundarias como fisuras, localizadas sobre cuello, miembros superiores, pliegues inframamarios y glúteos, sin evidencia de síntomas sistémicos (Ver Figura 1). Inicialmente se consideró un posible cuadro de psoriasis, dada las características eritemato-descamativas y la distribución topográfica de las lesiones. También se consideró un posible cuadro de pénfigo por las características de las lesiones y la localización en miembros superiores, motivo por el cual se le solicitó una biopsia de piel, que se tomó de la cara antero-lateral del cuello, que mostró acantolisis epidérmica y hendiduras intraepidérmicas por pérdida progresiva de los puentes intercelulares, dando el aspecto de ‘muro en ruinas’, sugestiva de enfermedad de PBF (Ver Figura 2). Con base en los hallazgos clínicos e histopatológicos se integró el diagnóstico de PBF y se descartaron otras dermatosis infrecuentes, como la enfermedad de Darier-White, la enfermedad de Grover o dermatosis acantolítica transitoria. No se realizó ningún otro examen incluyendo estudios genéticos, ni la inmunofluorescencia directa, la cual no se le había ordenado previamente según se revisó en la historia clínica. Se inicia manejo con ácido fusídico tópico y cefalexina oral para reducir la sobreinfección bacteriana y la fetidez asociada, asociado a prednisona 40 mg por 10 días. Una semana más tarde se dejó solamente el corticoide tópico dos veces al día, con mejoría progresiva de las lesiones durante 3 meses. Pasados 4 meses de iniciado el tratamiento, al examen físico reveló un compromiso del 2% de la superficie corporal con respecto al examen inicial, sin posterior exacerbación de síntomas, sin ingresos hospitalarios, sin efectos adversos de corticoterapia y una mejoría significativa en la calidad de vida.
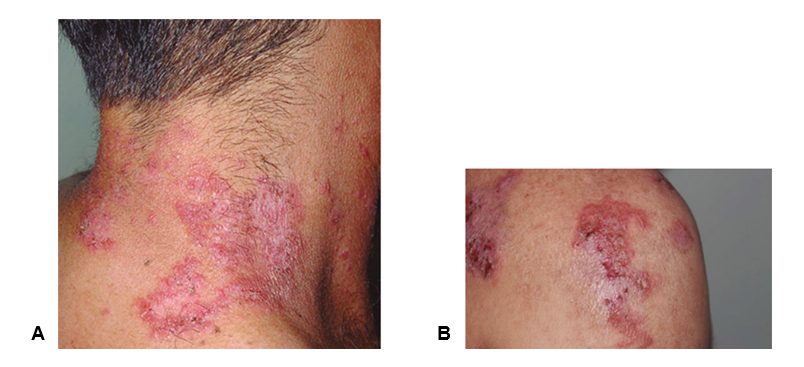
Fuente: autores
Figura 1 Placas circinadas de bordes bien definidos de tonalidad rosada con áreas descamativas y hematocostras Localizadas en la nuca y cara lateral de cuello también está presente en la región deltoidea derecha de un hombre de mediana edad
Discusión
El PBF se trata de una dermatosis de herencia autosómica dominante con penetrancia incompleta, expresividad variable y curso crónico, puede afectar a hombres y mujeres por igual, con presentación bimodal en la adolescencia tardía o en la edad adulta. Se presenta retraso en su diagnóstico, en especial si las lesiones responden a tratamientos tópicos con corticoides durante períodos cortos. Además de las lesiones en la piel, el PBF afecta el componente social del paciente, sin amenazar su supervivencia.
El defecto genético responsable del PFB se localiza en el cromosoma 3q21-q24, y se debe a mutaciones del gen ATP2C1 que codifica la proteína SPCA1, la cual constituye una bomba de calcio y magnesio del aparato de Golgi de la célula2-3-4. El calcio a su vez participa en la formación de las cadherinas (moléculas de adhesión intraepidérmica). Esta mutación genera un defecto generalizado en la adhesión de los queratinocitos (explica la acantolisis), que permanece normalmente subclínica y solo se pone de manifiesto tras determinados estímulos físicos externos como fricción, luz ultravioleta, quemadura7.
Más de dos tercios de los pacientes permanecen asintomáticos a lo largo de la vida1-2. Las lesiones están localizadas casi siempre en los pliegues (cuello, ingle y axila), y el curso de la enfermedad suele ser crónico, con el desarrollo de brotes que duran meses, seguido de períodos de remisión de la enfermedad4. La fricción, el calor o el sudor exacerban la sintomatología8-9. La morfología de las lesiones es poco específica, y en ausencia de un alto índice de sospecha de la enfermedad el diagnóstico es casi siempre incorrecto, siendo frecuente hacer diagnóstico de eccema, micosis o infección bacteriana. La lesión primaria es una placa eritematosa de bordes irregulares que se extiende por toda la superficie corporal y puede contener en su interior vesículas, pústulas y costras. Desde el punto de vista histopatológico se evidencian hendiduras intraepidermicas y acantolisis suprabasal extensa, que afecta el estrato espinoso, dando una imagen en «pared de ladrillos en ruinas» por la pérdida masiva de los puentes celulares entre queratinocitos11-12.
Es indispensable diferenciar las dermatosis que cursan con un rash pruriginoso en las áreas intertriginosas, como son la dermatitis de contacto, la dermatitis atópica, la tinea cruris, el pénfigo vulgar y vegetante, la psoriasis invertida e intertrigos de diversas etiologías13. Las lesiones que sean blanquecinas sobre un fondo erosivo hacen pensar en intertrigos candidiásicos. El examen micológico puede desorientar al clínico al mostrar la presencia de levaduras y pseudomicelios, pero estas lesiones no desaparecen con el tratamiento antifúngico exclusivo, ya que en ocasiones coexisten en el enfermo. El aspecto circinado con bordes eritemato-escamosos y tendencia a la cicatrización central puede semejar una dermatoficia, en este caso, el examen micológico es siempre negativo. Las lesiones costrosas y exudativas semejan un impétigo14. Los pacientes tienen un riesgo aumentado para desarrollar dermatitis de contacto alérgico a los medicamentos tópicos utilizados. Esto se debe a que es una enfermedad crónica, que requiere tratamientos prolongados con preparaciones tópicas y que la función de la barrera cutánea se encuentra alterada. La localización en el cuero cabelludo plantea el diagnóstico diferencial con la dermatitis seborreica, si bien la misma puede ser un factor desencadenante15. Desde el punto de vista histológico, los diagnósticos diferenciales se plantean con dermatosis que presentan acantolisis suprabasal y disqueratosis, como el pénfigo vulgar, la enfermedad de Darier-White y la enfermedad de Grover o dermatosis acantolítica transitoria.
El tratamiento está dirigido a reducir la inflamación y prevenir los brotes. Aún no existe un protocolo claro para el tratamiento de esta enfermedad16. Varias opciones de tratamiento incluyen corticosteroides tópicos y sistémicos, 5- fluorouracilo tópico, análogos tópicos de vitamina D, óxido de zinc tópico, dapsona, psoraleno más ultravioleta A, retinoides sistémicos, ciclosporina, metotrexato y terapia fotodinámica17. Se ha demostrado que el ácido Oxiding in vitro e in vivo posee propiedades que retardan el estrés oxidativo, mejorando la reparación de las heridas18. Reciente evidencia sugiere que la naltrexona oral en dosis bajas es efectiva para el PBF, pero su limitación es la recaída inmediata después de la interrupción de la medicación19. El tratamiento con corticosteroides tópicos y sistémicos como el empleado en nuestro caso, busca reducir la inflamación. También se han informado casos con buena respuesta al manejo con ciclosporina17, metotrexato, y talidomida20, lo que sugiere que los fármacos inmunomoduladores podrían ser útiles para casos graves. Las formas más graves de PBF son las de más difícil manejo.
El Apremilast inhibe específicamente la actividad de la fosfodiesterasa 4 de adenosin monofosfato cíclico disminuyendo la activación de TH1 y TH175. En una serie de casos de cuatro pacientes con PBF severa mostraron buena respuesta tras el empleo de Apremilast con mejora significativa de las lesiones representada por marcada reducción en el número y la intensidad de sus brotes en un período de seguimiento de 6 a 10 meses. En los casos de recurrencias los pacientes obtenían buena respuesta con el empleo de corticoides tópicos20.
Conclusiones
El PBF es una enfermedad que impacta la calidad de vida de los que la padecen que además del reconocimiento oportuno y manejo médico, requiere de educación en el paciente sobre su enfermedad, sus posibles desencadenantes o precipitantes, cómo evitar y controlar los brotes, siendo de gran importancia la educación y sensibilización de esta entidad y su manejo en los profesionales médicos. Nuestro paciente antes de ser referido a nosotros, usó una serie de tratamientos tópicos por sospecha diagnóstica de psoriasis, dermatitis de contacto, y micosis superficial, siendo estos poco efectivos para su condición. No obstante, posterior a obtener el resultado del estudio histopatológico de piel, permitió hacer un correcto diagnóstico e indicar corticoides sistémicos en el plan de tratamiento, con resolución y remisión de las lesiones. Dada la poca evidencia del PBF y la adecuada evolución del paciente del caso clínico, consideramos este caso puede ser de ayuda en el manejo del PBF, siendo necesario el desarrollo de evidencia más sólida en el manejo de estos pacientes.

