











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El síndrome de Camurati-Engelmann, también conocido como displasia diafisaria progresiva, es una enfermedad rara, autosómica dominante, con penetrancia incompleta y expresividad variable1, que se caracteriza por aumento de la masa ósea haciendo parte del grupo de las displasias óseas esclerosantes2,3. Fue descrita por primera vez en 1920 por Cockayne en un niño de 9 años, quien consideró estos hallazgos como una manifestación anormal de sífilis congénita4. En 1922, Camurati planteó este síndrome como una enfermedad hereditaria5 y, en 1929, Engelmann reportó otro caso cuya manifestación fue atrofia muscular y marcado compromiso óseo6. El nombre de displasia diafisaria progresiva fue propuesto por Neuhauser en 1948, debido al compromiso diafisario constante y la presentación gradual de la hiperostosis7, sin embargo, el epónimo de Camurati-Engelmann es ampliamente aceptado.
La información epidemiológica conocida de esta enfermedad no es muy amplia, pero se estima que su prevalencia sea de uno por cada millón de habitantes. No se han observado diferencias significativas en su distribución por sexo o raza8. Si se considera el reporte de 24 familias de América, Europa, África y Oceanía, se puede afirmar que la distribución de esta patología es global, sin predominar en alguna región geográfica específica9. En total se ha identificado en, al menos, 300 individuos10.
La variabilidad observada en los casos familiares reportados es típica de los desórdenes dominantes. A pesar de que no se conoce con exactitud la razón de dicha variabilidad, algunas hipótesis deberían ponerse en consideración para explicar este fenómeno: múltiples isoalelos normales, múltiples alelos mutantes en el mismo locus que causan la enfermedad, la presencia de diferentes modificadores no alélicos o modificadores ambientales1.
El gen responsable de la displasia diafisaria progresiva fue localizado recientemente a nivel de la región 19q13.1-13.311, este gen tiene siete exones y la mayoría de dichas mutaciones están en el exón 4, seguido del 1. Así mismo, los tres alelos mutantes más frecuentes son p. Arg210cys, p. Arg218His y p. Cys225Arg7. Se conoce que codifica el factor de crecimiento beta 1 (TGF-β1), una proteína polipeptídica que actúa en todo tipo de células, pero cuyo mayor repositorio es el hueso, y muchas células óseas expresan receptores para TGF-β1. Se ha reportado la presencia de variantes genéticas patogénicas en dicho gen, lo cual puede ser también un factor que contribuya a la alta variabilidad relacionada con el síndrome de Camurati-Engelmann12.
Datos provenientes de múltiples experimentos in vitro han demostrado el rol del TGF-β1 en cada etapa de la formación ósea. Incrementa la formación ósea in vitro por medio del reclutamiento de progenitores osteoblásticos y estimula su proliferación, aumentando de esta manera el conjunto de osteoblastos involucrados y promoviendo las etapas tempranas de diferenciación (producción de matriz ósea)13. El TGF-β1 también bloquea la apoptosis de los osteoblastos al mantener la supervivencia durante la transdiferenciación en osteocitos14,15,16, y tiene una acción inhibitoria en la miogénesis y la adipogénesis, lo que explicaría los síntomas no óseos de la enfermedad17,18.
Lo anterioriormente descrito demuestra el importante papel de esta proteína como principal reguladora de la función osteoblástica y osteoclástica en el tejido esquelético. Al ocurrir la mutación en esta patología, se produce un desequilibrio entre la deposición esquelética y la reabsorción. Este desequilibrio resulta a su vez en hiperostosis, que afecta principalmente las diáfisis de los huesos largos. A medida que progresa la enfermedad, se afectan también las metáfisis y pueden desarrollarse cambios escleróticos en el cráneo o la pelvis. Las deformidades esqueléticas que aparecen con el tiempo son las responsables de la mayor parte de los signos y síntomas de la enfermedad19,20,21, y traen como consecuencia dolor en las extremidades, debilidad muscular proximal, atrofia muscular, marcha atáxica, fatigabilidad y cefalea22.
El inicio de esta enfermedad suele ocurrir durante la infancia, entre los 4 y 10 años, progresa en la adolescencia y queda estacionaria o avanza lentamente en la edad adulta. Los síntomas clínicos están presentes en más de 74% de los enfermos18. La enfermedad puede iniciar con la aparición de marcha claudicante en el 64% de las personas, dolor óseo en el 90%, fatiga en un 43% y contracturas articulares en el 43%. Adicionalmente, se presenta atrofia muscular progresiva y disminución de la grasa subcutánea en las extremidades, se puede observar un hábito corporal marfanoide y estatura en percentiles altos para la edad, macrocefalia con frente prominente, lordosis o escoliosis dorsal, pie plano, prognatismo y compromiso de bulbo raquídeo, amígdala y pares craneales (especialmente el VI, VII y VIII), u otras alteraciones del sistema nervioso, como clonus y ataxia cerebelosa, dado que también se puede hallar compromiso óseo en los orificios craneales8. También es frecuente encontrar compromiso hormonal, manifestado por pubertad tardía con hipogonadismo secundario y crecimiento óseo prolongado9. En otras ocasiones se pueden encontrar hepatoesplenomegalia, fenómeno de Raynaud, hiperhidrosis de manos y pies, y retraso en la dentición23.
Al analizar los hallazgos de laboratorio, se puede observar anemia, leucopenia, aumento en la velocidad de sedimentación globular, fosfatasa alcalina, paratohormona, calcio y fósforo sérico9, todo lo cual sugiere que el síndrome de Camurati-Engelmann no solo es una patología ósea, sino que sus manifestaciones también son sistémicas.
El diagnóstico se obtiene con base en hallazgos clínicos y radiológicos característicos de esta enfermedad, con confirmación genética. El examen físico se enfoca en la búsqueda de alteraciones neurológicas y osteomusculares, dado el compromiso sistémico de la enfermedad y, en los estudios radiológicos pertinentes, el hallazgo de un engrosamiento cortical en la diáfisis de aspecto fusiforme, que puede extenderse hasta las metáfisis (sin afectar las epífisis), además, la proliferación del tejido óseo en el periostio y endostio que lleva a un estrechamiento del canal medular24,25,26. Los huesos afectados en orden de frecuencia son: fémur, tibia, peroné, húmero, cúbito y radio; puede observarse compromiso de la mandíbula, escápulas, clavículas, pelvis y base de cráneo, rara vez existe compromiso de huesos del carpo, tarso y falanges. La imagen radiológica típica en huesos largos se observa en el 94% de los pacientes, un 54%, además, puede tener manifestaciones craneales y un 63% en la pelvis9.
A continuación, se presenta el caso de una paciente de 33 años con síndrome de Camurati-Engelmann confirmado por secuenciación exómica, caracterizado por distonías y un deterioro progresivo del estado general. Asimismo, se exponen generalidades terapéuticas y nuevas modalidades de tratamiento en estudio.
Presentación del caso
Paciente femenina de 33 años, producto del primer embarazo de padres no consanguíneos sin historia familiar de enfermedades, procedente de Manizales, quien consultó en febrero de 2014 por movimientos anormales presentes desde 2006, reconocidos como distonías de predominio craneocervical. En seguimiento por neurología, recibía tratamiento con toxina botulínica a una dosis de 250 unidades intramusculares guiada con electromiografía, con mejoría parcial. En diciembre de 2016, presentó una crisis distónica, con compromiso de cabeza, cuello y miembros inferiores que cesaba durante el sueño, sin un desencadenante claro.
Fue valorada en una institución de cuarto nivel de la ciudad, donde indican manejo ambulatorio con biperideno en un esquema de 2 mg vía oral cada 24 horas durante la primera semana, cada 12 horas durante la segunda semana, y cada 8 horas durante la tercera y cuarta semana. La paciente presentó mejoría parcial inicial, pero en noviembre de 2017 aparecieron además disfagia y disartria, por lo cual fue valorada en una institución de la ciudad donde se descartaron patologías estructurales y metabólicas relacionadas con compromiso del Sistema Nervioso Central. Para este momento se sospechó enfermedad de Huntington y se decidió realizar estudio genético, que finalmente detectó la presencia de dos alelos de 16 y 22 repeticiones en el gen HTT, por lo que se descartó dicha patología.
En septiembre de 2018 se hizo una prueba de secuenciación del exoma completo, utilizando la tecnología Illumina a partir de sangre venosa, donde se realizó evaluación de diferentes genes relacionados con las distonías aisladas y combinadas y no se encontraron asociaciones, sin embargo, se reportó una variante patogénica heterocigota en TGF-β1, siendo p. Arg156Cys, en el exón 2, el alelo mutante. Con el software de predicción se estableció un significado clínico patogénico de la variante identificada y compatible con el diagnóstico genético de la enfermedad de Camurati-Engelmann. Un reporte de radiografía evidenció engrosamiento de las corticales diafisarias en extremidades superiores e inferiores.
En marzo de 2019 reconsultó por empeoramiento de los síntomas previos, deterioro funcional y aumento de los movimientos distónicos, dado por movimientos generalizados coreoatetósicos que se exacerbaron constantemente, sin alteraciones de la conciencia o relajación de esfínteres. Se realizó manejo con 15 mg de midazolam cada 4 horas, sin obtener respuesta adecuada. Al examen físico se evidenció atrofia y retracciones musculares generalizadas, palidez de piel, macrocefalia, frente prominente, extremidades con huesos gruesos y arqueados, debilidad, reflejos disminuidos y leve rigidez generalizada, pie plano, hiperhidrosis en manos y pies, y manifestación de dolor óseo. Los exámenes complementarios mostraron anemia y, en la radiografía de tórax, infiltrados de tipo no específico en ambos hemitórax y escoliosis dorsal de convexidad derecha, sin otras alteraciones.
Durante su estancia hospitalaria se evidenció declinación funcional progresiva, hasta llegar a presentar dependencia total para sus actividades de vida diaria, además, empeoramiento de movimientos anormales, dolor óseo persistente, aumento de la atrofia muscular y caquexia refractaria. En el seguimiento realizado por genética se consideró que lo anterior era producto de la progresión crónica, irreversible y falta de cura del síndrome de Camurati-Engelmann, por lo que se solicitó valoración y manejo por cuidado paliativo. Se consideró entonces que la paciente y familia requerían intervenciones terapéuticas con orientación paliativa, teniendo en cuenta que, a este momento, se trataba de una enfermedad avanzada, incurable, progresiva, con presencia de síntomas multifactoriales, sin posibilidades razonables de respuesta al manejo actual y sin existir en el momento un tratamiento específico.
Discusión
En este reporte de caso, se realizó un análisis tanto clínico como paraclínico del síndrome de Camurati-Engelmann. Es de resaltar que la paciente cumplía con criterios clínicos, como el compromiso osteomuscular, atrofia, dolor óseo, anemia y pubertad tardía además, como criterios radiográficos se hallaron hiperostosis diafisometafisaria en los huesos largos de la paciente18. Todo esto en concordancia con el diagnóstico genético del síndrome de Camurati-Engelmann.
En comparación con los dos casos descritos del síndrome en el país, los tres pacientes han manifestado dolor óseo en miembros superiores e inferiores, el cual es crónico, inespecífico, sin irradiación, que se puede agudizar en la noche y no cede con AINES. En los tres casos se mostró engrosamiento de las corticales diafisiarias con pérdida del canal medular. En los dos casos anteriores no se hace referencia a las demás manifestaciones clínicas características de la enfermedad, los estudios moleculares realizados, ni manifiestan algún tipo de compromiso neurológico como el que se dio en la paciente del presente caso9,18.
La baja incidencia y la alta variabilidad de esta enfermedad llevan con frecuencia a que pase inadvertido su diagnóstico, debido a la presencia de manifestaciones inespecíficas o hallazgos incidentales en radiografías, que llevan al personal médico a la decisión de tomar exámenes e iniciar conductas terapéuticas erradas ante la sospecha de otras patologías relacionadas con el sistema musculoesquelético22. Es por esto que se debe hacer un adecuado enfoque de diagnóstico y diferencial, y examen histopatológico y genético, relevante, considerando principalmente el grupo de las displasias óseas esclerosantes27.
Se ha postulado que dicha variabilidad se debe a fenómenos genéticos previamente mencionados, como isoalelos normales, alelos mutantes en el mismo locus, y modificadores no alélicos y ambientales. Recientemente ha cobrado especial importancia la existencia de mutaciones en el gen que codifica la proteína TGF-β1. Campos-Xavier et al., en un estudio llevado a cabo en 2001, demostraron que la presencia de un alelo T en la posición -509 pb se asoció con concentraciones plasmáticas más altas de TGF-β1, y esto aplica particularmente para los individuos homocigóticos del alelo T en comparación con los heterocigotos, lo que sugiere un efecto dosis-respuesta de la sustitución de T sobre las concentraciones circulantes de TGF-β1, con la consecuente hipótesis de que las variantes patogénicas de TGF-β1 podrían alterar el nivel de expresión génica y la gravedad de los síntomas clínicos. Estos autores llegaron a la conclusión de que el síndrome de Camurati-Engelmann es una condición con variable presentación clínica que se debe a mutaciones parcialmente penetrantes del gen TGF-β1. Esta variación en la expresión clínica no es debido a un nivel variable de expresión del gen TGF-β1, pero puede ser en parte explicada por la presencia de genes modificadores28. La variante patogénica encontrada consiste en una transición de citosina a timina en la posición 466, la cual modifica en la proteína el aminoácido arginina por cisteína en la posición 156, que generó la afectación del TGF-β1.
Hughes y colaboradores propusieron, en su estudio de 10 miembros de una familia, en quienes se encontró la variante R218H en el gen TGF-β1, que la resolución espontánea observada en algunos de estos individuos puede explicar en parte la amplia variabilidad fenotípica. Parece ser que el inicio en la infancia temprana se asocia con un fenotipo más severo, mientras que el inicio en la adolescencia tiene una mayor probabilidad de lograr la remisión espontánea29. De acuerdo con lo anterior, se considera que la severidad del curso clínico de la paciente y su fallecimiento temprano se asocian con un inicio en la infancia y un fenotipo de mayor gravedad.
El hallazgo y curso de las distonías vistas en esta paciente parecen ser una manifestación inusual de la enfermedad puesto que, hasta la fecha, no se ha encontrado reporte en la literatura internacional de una presentación similar. A nivel neurológico, las manifestaciones que se han reportado incluyen afecciones visuales y otológicas. Kim et al documentaron, en una serie de casos publicada en 2018, dos pacientes con exoftalmos y uno de ellos con visión borrosa a los 29 años y desprendimiento de retina en el ojo derecho; además de un tercer paciente con mareos que ocurrían varias veces al día, en el cual se encontró que tenía vestibulopatía recurrente en el examen otológico50. Meczekalski et al reportaron el caso de una paciente de 21 años con amenorrea hipotalámica, quien cursó además con sordera total del oído izquierdo e hipoacusia conductiva en el oído derecho, documentadas al examen audiológico y relacionadas con el hallazgo radiológico de engrosamiento generalizado y difuso, y aumento de la esclerosis de casi todos los huesos craneales visibles, así como una reconstrucción osteosclerótica significativa de las estructuras del oído interno, alteraciones identificadas en la tomografía axial computarizada de huesos temporales21. Owhonda y colaboradores, por su parte, documentaron el caso de una paciente de 44 años, que manifestó fatiga, rigidez muscular, exoftalmos, mareos y diplopía, cuyo único hallazgo a la resonancia magnética cerebral fue el engrosamiento de la base del cráneo51. Este tipo de manifestaciones neurológicas pueden ser explicadas por la destrucción de vías neurales producto de la compresión de partes particulares del sistema nervioso por el hueso hipertrófico, especialmente aquellas relacionadas con la modalidad auditiva o visual, y la pérdida de audición puede también ser el resultado de la oclusión de las estructuras del hueso temporal del oído medio o interno. A pesar de este conocimiento, no se ha descrito, hasta el momento, un modelo fisiopatológico claro que explique la génesis de las distonías que fueron presentadas por la paciente del caso actual, lo cual hace que el presente reporte de caso sea de especial interés y pueda abrir las puertas a futuras investigaciones que permitan una comprensión más amplia de esta patología.
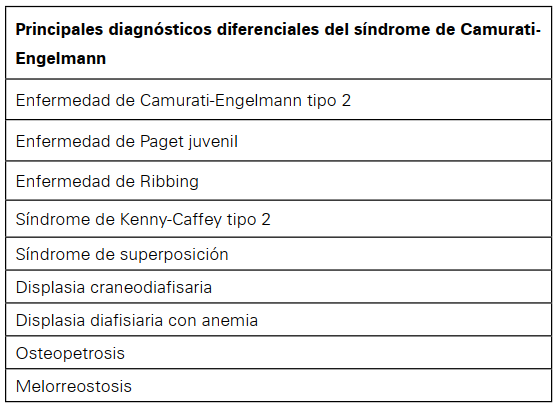
Para el diagnóstico diferencial del síndrome de Camurati-Engelmann se deben considerar las enfermedades que causan afectación esclerótica del tejido óseo, que se exponen a continuación: (Ver Tabla 1)
Tabla 1 Principales diagnósticos diferenciales del síndrome de Camurati-Engelmann.
Fuente: Tomado y adaptado de: Restrepo JP, Molina MP. Enfermedad de Camurati-Engelmann: Reporte de un caso y revisión de la literatura. Rev Colomb Reumatol. 2016;23(3):218–222.
Otras patologías importantes para considerar son osteomielitis esclerosante crónica, periostitis multifocal, hiperostosis inducida por prostaglandina E1, fluorosis crónica y osteosclerosis intramedular32.
Aún no se conoce un tratamiento específico para el síndrome de Camurati-Engelmann, sin embargo, el uso de glucocorticoides tiene un efecto positivo dado por sus propiedades antiinflamatorias e inmunosupresoras que conlleva a mejorar algunos de los síntomas (dolor, debilidad muscular, fatiga). Es preciso mencionar que no influyen en la progresión del síndrome13, aunque se ha documentado que los esteroides podrían aumentar la resorción ósea y disminuir su depósito laminar38,39. A los pacientes con síntomas leves se les puede administrar 0.5 mg a 1 mg/kg de prednisolona en días alternos pudiendo interrumpirse por periodos. A los pacientes con síntomas graves se les indica administrar un bolo de 1 a 2 mg/kg/día, seguido de la dosis más baja tolerada en días alternos, también pueden utilizarse dosis altas en crisis de dolor. Es importante mantener la dosis más baja eficaz con el fin de prevenir los efectos secundarios de los corticoides. El losartán puede ser utilizado en pacientes sintomáticos que no toleran los corticoides, pues se ha encontrado que tiene efecto regulador en el proceso de señalización de TGF-β140. En algunos pacientes se ha evidenciado reducción del dolor y aumento de la fuerza muscular, pero no hay evidencia suficiente que respalde el éxito de dicha terapia41.
Existen pocos registros del uso de bifosfonatos y calcitonina para el tratamiento del síndrome de Camurati-Engelmann, a pesar de que en teoría podrían ser útiles42,43,44.
La cirugía está indicada para el tratamiento de la hiperostosis craneal que ocasione hipertensión intracraneal, con reporte de buenos resultados45. También se han realizado osteotomías y limpieza del canal medular como tratamiento de las hiperostosis de huesos largos46 y descompresión quirúrgica del conducto auditivo interno en aquellos casos de sordera con mejoría de los síntomas, pero estos tratamientos son controvertidos ya que estas lesiones pueden recurrir por la progresión de la enfermedad47.
Teniendo en cuenta el papel central que cumple el TGF-β1 en la patogenia del síndrome de Camurati-Engelmann, se ha considerado que la inhibición de los receptores de esta proteína puede tener un efecto benéfico en la progresión de la enfermedad. Sin embargo, la administración sistémica de estos inhibidores puede tener efectos secundarios graves en la respuesta inmunitaria y el proceso de angiogénesis, debido a que este receptor está ampliamente expresado en todo el organismo48. En un estudio reciente de Qin et al., se diseñó un conjugado alendronato-TβR1I unido por un enlace escindible que permite que el conjugado se una al hueso primero y luego se libere el TβR1I gradualmente. El conjugado atenuó eficazmente la progresión del síndrome a una dosis de 100 mcg/kg por semana. La dosis total de TβR1I se redujo 700 veces después de dicha modificación de focalización ósea y la dosis efectiva osciló entre 100 mcg/kg y 1 mg/kg por semana, lo que respalda el uso de varias dosis diferentes en futuras aplicaciones clínicas49.
Hasta el momento, el pronóstico del síndrome de Camurati-Engelmann sigue siendo sombrío, dada la ausencia de un tratamiento eficaz y la tendencia a la progresión de las complicaciones con el compromiso de la calidad de vida de los pacientes afectados..
Una vez hecho el diagnóstico de la enfermedad, con base en sus manifestaciones clínicas y alteraciones radiológicas, es importante realizar un estudio genético con el fin de determinar si existe la mutación. Hay pacientes con presencia de mutación y sin manifestaciones clínicas, es decir, con penetrancia incompleta, de igual forma existen pacientes con las manifestaciones y sin presencia de la mutación. Por consiguiente, se recomienda un estudio genético y de imágenes diagnósticas a progenitores con el fin de estimar el riesgo hereditario y proporcionar consejería genética..
Enfermedad de Camurati-Engelmann tipo 2. Esta entidad simula clínica y radiológicamente el síndrome de Camurati-Engelmann, sin embargo, en esta no se encuentra la mutación en el gen TGF-β124.
Enfermedad de Paget juvenil. En ella se encuentra expansión diafisaria de los huesos tubulares largos, esclerosis craneal y aumento de la fosfatasa alcalina sérica, pero en esta los huesos son más arqueados, hay trabeculación irregular y desmineralización generalizada30.
Enfermedad de Ribbing. Radiológicamente es indistinguible del Síndrome de Camurati-Engelmann, también es causada por mutaciones del TGF-β1, por lo que se considera que ambas son variables fenotípicas del mismo trastorno, pero algunos autores estiman que, por ser la Enfermedad de Ribbing de herencia autosómica recesiva, se trata de una etiología separada26. También es importante destacar que en el Síndrome de Camurati-Engelmann la afectación es bilateral y simétrica, mientras que en la enfermedad de Ribbing suele presentarse de forma unilateral o bilateral con afectación asimétrica31.
Síndrome de Kenny-Caffey tipo 2. Es una displasia de osificación intramembranosa en la cual hay engrosamiento cortical de los huesos largos, pero se presenta retraso en el cierre de las fontanelas, hipocalcemia e hipoparatiroidismo, afectaciones que están ausentes en el síndrome de Camurati-Engelmann8.
Síndrome de superposición. Es la combinación de dos o más displasias esclerosantes que suman sus manifestaciones clínicas, por lo cual se debe tener en cuenta que las displasias no son entidades distintas y pueden existir factores comunes en su patogenia y herencia32.
Displasia craneodiafisaria. En ella, la esclerosis del cráneo y los huesos faciales es más pronunciada, hay ensanchamiento de las costillas, clavículas y huesos tubulares cortos de las manos, la afectación de los huesos largos se restringe a la diáfisis, características que no hacen parte del Síndrome de Camurati-Engelmann33.
Displasia diafisaria con anemia. En general la hiperostosis es menos pronunciada, hay anemia severa y mayor susceptibilidad a infecciones. No hay evidencia de formación ósea en el endostio ni subperiostio, lo que sí ocurre en el síndrome de Camurati-Engelmann34.
Osteopetrosis. En ella se encuentra que la diáfisis de los huesos tubulares largos es de forma normal, y el modelado defectuoso se refleja en el ensanchamiento de las regiones sub metafisarias de los huesos tubulares, a diferencia del síndrome de Camurati-Engelmann35.
Melorreostosis. En la entidad se encuentra hiperostosis de huesos largos con afectación asimétrica y unilateral que compromete uno o a varios huesos de una sola extremidad36 y la formación de hueso es más pronunciada37.
Otras patologías importantes para considerar son osteomielitis esclerosante crónica, periostitis multifocal, hiperostosis inducida por prostaglandina E1, fluorosis crónica y osteosclerosis intramedular32.
Aún no se conoce un tratamiento específico para el síndrome de Camurati-Engelmann, sin embargo, el uso de glucocorticoides tiene un efecto positivo dado por sus propiedades antiinflamatorias e inmunosupresoras que conlleva a mejorar algunos de los síntomas (dolor, debilidad muscular, fatiga). Es preciso mencionar que no influyen en la progresión del síndrome13, aunque se ha documentado que los esteroides podrían aumentar la resorción ósea y disminuir su depósito laminar38,39. A los pacientes con síntomas leves se les puede administrar 0.5 mg a 1 mg/kg de prednisolona en días alternos pudiendo interrumpirse por periodos. A los pacientes con síntomas graves se les indica administrar un bolo de 1 a 2 mg/kg/día, seguido de la dosis más baja tolerada en días alternos, también pueden utilizarse dosis altas en crisis de dolor. Es importante mantener la dosis más baja eficaz con el fin de prevenir los efectos secundarios de los corticoides. El losartán puede ser utilizado en pacientes sintomáticos que no toleran los corticoides, pues se ha encontrado que tiene efecto regulador en el proceso de señalización de TGF-β140. En algunos pacientes se ha evidenciado reducción del dolor y aumento de la fuerza muscular, pero no hay evidencia suficiente que respalde el éxito de dicha terapia41.
Existen pocos registros del uso de bifosfonatos y calcitonina para el tratamiento del síndrome de Camurati-Engelmann, a pesar de que en teoría podrían ser útiles42,43,44.
La cirugía está indicada para el tratamiento de la hiperostosis craneal que ocasione hipertensión intracraneal, con reporte de buenos resultados45. También se han realizado osteotomías y limpieza del canal medular como tratamiento de las hiperostosis de huesos largos46 y descompresión quirúrgica del conducto auditivo interno en aquellos casos de sordera con mejoría de los síntomas, pero estos tratamientos son controvertidos ya que estas lesiones pueden recurrir por la progresión de la enfermedad47.
Teniendo en cuenta el papel central que cumple el TGF-β1 en la patogenia del síndrome de Camurati-Engelmann, se ha considerado que la inhibición de los receptores de esta proteína puede tener un efecto benéfico en la progresión de la enfermedad. Sin embargo, la administración sistémica de estos inhibidores puede tener efectos secundarios graves en la respuesta inmunitaria y el proceso de angiogénesis, debido a que este receptor está ampliamente expresado en todo el organismo48. En un estudio reciente de Qin et al., se diseñó un conjugado alendronato-TβR1I unido por un enlace escindible que permite que el conjugado se una al hueso primero y luego se libere el TβR1I gradualmente. El conjugado atenuó eficazmente la progresión del síndrome a una dosis de 100 mcg/kg por semana. La dosis total de TβR1I se redujo 700 veces después de dicha modificación de focalización ósea y la dosis efectiva osciló entre 100 mcg/kg y 1 mg/kg por semana, lo que respalda el uso de varias dosis diferentes en futuras aplicaciones clínicas49.
Hasta el momento, el pronóstico del síndrome de Camurati-Engelmann sigue siendo sombrío, dada la ausencia de un tratamiento eficaz y la tendencia a la progresión de las complicaciones con el compromiso de la calidad de vida de los pacientes afectados8.
Una vez hecho el diagnóstico de la enfermedad, con base en sus manifestaciones clínicas y alteraciones radiológicas, es importante realizar un estudio genético con el fin de determinar si existe la mutación. Hay pacientes con presencia de mutación y sin manifestaciones clínicas, es decir, con penetrancia incompleta, de igual forma existen pacientes con las manifestaciones y sin presencia de la mutación. Por consiguiente, se recomienda un estudio genético y de imágenes diagnósticas a progenitores con el fin de estimar el riesgo hereditario y proporcionar consejería genética8.
Conclusiones
El síndrome de Camurati-Engelmann es una displasia ósea esclerosante rara, autosómica dominante, con penetrancia incompleta y expresividad variable. Son escasos los reportes de caso, y esto, en conjunto con la variabilidad en las manifestaciones clínicas y en su curso, hace que se conozca poco sobre esta patología y las opciones de un manejo definitivo. Su tratamiento, hasta el momento, se basa en el control sintomático, pero las investigaciones recientes relacionadas con las bases genéticas obligan a una búsqueda de tratamientos que puedan impactar favorablemente el curso de la misma. Se espera que este nuevo reporte de caso permita a los médicos pensar en el síndrome de Camurati-Engelmann como una alternativa diagnóstica en pacientes con enfermedades que causan afectación esclerótica del tejido óseo y a futuro se generen más investigaciones en torno a su tratamiento.
Consideraciones éticas
Se contó con el consentimiento informado debidamente diligenciado por la acudiente de la paciente, en presencia de la misma.
Limitaciones
Dentro de las limitaciones para el desarrollo de este artículo, destaca la ausencia de las imágenes correspondientes al estudio radiológico de huesos largos hecho a la paciente, cuya importancia para ilustrar adecuadamente el caso los autores reconocemos. También, es pertinente resaltar que no fue posible contar con la asesoría de los profesionales en genética que atendieron este caso en la institución respectiva.

