











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Las miopatías inflamatorias constituyen el principal grupo de causas adquiridas de debilidad muscular, estas se clasifican en: polimiositis, dermatomiositis y miositis con cuerpos de inclusión1. La polimiositis se caracteriza por debilidad muscular proximal y simétrica2, se debe realizar diagnóstico diferencial con distrofias musculares, miopatías metabólicas, endocrinas, enfermedades de motoneurona o unión mioneural, miopatía por esteroides e intoxicación por estatinas3.
Su principal mecanismo fisiopatológico es la infiltración de linfocitos TCD8 y macrófagos en las fibras musculares, que conducen a su lisis2. Además puede afectar músculos respiratorios, músculos de la deglución y el miocardio. Los cambios clínicos son debidos a la inflamación crónica de la musculatura estriada4. Cursa con elevación de la concentración plasmática de diferentes enzimas, entre ellas la fosfocreatincinasa (CPK), esencial en la actividad muscular para catabolizar la transferencia reversible del fosfato. La CPK escapa de un músculo deteriorado, por lo que su elevación indica destrucción muscular5.
La polimiositis se presenta entre los 45-64 años con una incidencia anual a nivel mundial de 4 casos por millón de habitantes3, siendo más frecuente en las mujeres con una relación de 2,5:1 casos frente a los hombres6y con reportes en Medellín, Colombia de 4:17, considerándose una entidad rara. La etiopatogenia es desconocida, aunque se considera un desorden autoinmune que involucra mecanismos hormonales, ambientales y mimetismos moleculares, genéticos o infecciosos8,9. Por otro lado, para su diagnóstico tradicionalmente se han utilizado los criterios de Bohan y Peter elaborados en 197510, que tienen sensibilidad entre 74-100% y especificidad de 29%11; sin embargo, recientemente el International Myositis Classification Criteria Project (IMCCP) publicó nuevos criterios de clasificación (Tabla 1), con sensibilidad del 93% y especificidad del 88%12, usadas para el diagnóstico y diferenciación de las miopatías inflamatorias según las características del paciente y los paraclínicos13.
Tabla 1 Criterios de clasificación de miopatías inflamatorias idiopáticas del International Myositis Classification Criteria Project (IMCCP)13.
| Edad de inicio (años): 0 - 17; 18 - 39 o ≥40 |
|---|
| Debilidad simétrica objetiva, generalmente progresiva, de las extremidades superiores proximales |
| Debilidad simétrica objetiva, generalmente progresiva, de las extremidades inferiores proximales |
| Flexores del cuello relativamente más débiles que los extensores |
| En las piernas, los músculos proximales son relativamente más débiles que los músculos distales |
| Erupción de heliotropo |
| Pápulas de Gottron |
| Signo de Gottron |
| Disfagia o dismotilidad esofágica |
| Anticuerpos anti-Jo-1 positivos |
| Niveles séricos elevados de CPK o lactato deshidrogenasa (LDH) o aspartato aminotransferasa o alanina aminotransferasa |
| Infiltración endomisial de células mononucleares que rodean, pero no invaden, las miofibras |
| Infiltración perimisial y/o perivascular de células mononucleares |
| Atrofia perifascicular |
| Vacuolas bordeadas |
Fuente: Unit of Biostatistics, Karolinska Institutet, Stockholm, Sweden. Classification Criteria for Idiopathic Inflammatory Myopathies. Disponible en: http://www.imm.ki.se/biostatistics/calculators/iim/
La miositis asociada al cáncer abarca cerca del 8% de los casos, con incidencia mayor a los 40 y 60 años. Puede preceder el comienzo del cuadro o aparecer 2 años después; se considera que un año o más reduce la asociación14-16. La tasa de mortalidad en mayores de 64 años es de 47.8% y 9.1% en pacientes más jóvenes. El sexo masculino, raza no caucásica, cáncer, compromiso esofágico, afectación respiratoria y disfunción cardíaca son predictores de mal pronóstico17.
En el tratamiento se deben considerar los efectos secundarios a largo plazo de la medicación17, también es importante la detección temprana de síntomas extramusculares como pérdida de peso, artralgias, fenómeno de Raynaud, compromiso digestivo, cardiaco, pulmonar o síntomas constitucionales, además de verificar la edad del paciente, grado de discapacidad, tolerancia, experiencia del médico y estado de salud general. El inicio del manejo es empírico y secuencial puede iniciar con prednisona en dosis altas, luego asociarse a azatioprina, micofenolato o metotrexato para un efecto con ahorro de esteroides, seguido de inmunoglobulina G intravenosa y por último un ciclo con rituximab, ciclosporina, ciclofosfamida o tacrolimus. Los pacientes que logran la recuperación funcional completa permanecen con tratamiento de mantenimiento1.
La sobrevida a cinco y diez años es cercana al 95 y 84% respectivamente, la muerte suele deberse a otras complicaciones por el deterioro sistémico y muscular al que lleva la enfermedad, principalmente pulmonares en el primer año y cardiacas en los cinco años siguientes al diagnóstico11,17, seguidas de infecciones (15%) y cáncer (11%)11, lo que refleja la importancia de un tratamiento temprano. El pronóstico es peor cuando la presentación debuta con afectación grave (disfagia severa o dificultades respiratorias) o cuando se retrasa el tratamiento1.
Al ser considerada una entidad rara, que es inusual su presentación en hombres, que no tiene un protocolo diagnóstico definido y que el manejo suele iniciar de manera empírica, puede representar un reto para el personal de salud y retrasar el tratamiento de la patología, que debe iniciar oportunamente para evitar el deterioro físico e incluso la muerte, por lo cual los objetivos del artículo fueron describir el manejo y seguimiento del caso de un paciente masculino con polimiositis, su agudización tres años después del diagnóstico y la comparación con el abordaje planteado en la literatura.
Caso clínico
En abril de 2016 un paciente masculino de 47 años de edad procedente de la zona urbana de Popayán, quien vive con su esposa y sus dos hijos, y se desempeña como guarda de seguridad, consulta a nivel I por debilidad muscular de 20 días de evolución, donde por posible accidente cerebro vascular es remitido al servicio de urgencias de nivel II. Al ingreso presentaba dolor poliarticular, disminución de movilidad y de fuerza en las cuatro extremidades de 20 días de evolución, con dolor a nivel de las caderas cuando cruza las piernas; refirió además estreñimiento en los últimos 15 días con cambio en el bolo fecal y pérdida de peso aproximado de 10 kg en 3 meses. Entre los antecedentes estaban migraña y dilatación de raíz aórtica, en manejo médico con atorvastatina y metoprolol; refiere consumo de bebidas alcohólicas ocasionalmente y niega consumo de otro tipo de sustancias, como antecedentes familiares refiere madre diabética. Se descartó accidente cerebrovascular al no presentar déficit motor, pudiendo incorporarse de la posición de cuclillas, elevar los miembros superiores, con fuerza prensil, marcha y nervios craneales normales, sin signos de irritación meníngea.
Se suspendió atorvastatina y los paraclínicos de ingreso reportaron CPK: 24000 U/L; hormona estimulante de tiroides (TSH): 10,91 U/L (4,5-10 UI/L) además de hipertransaminasemia. Se hospitalizó e inició levotiroxina 50mg/día y protocolo para descartar neoplasia colónica. Se indican paraclínicos para realizar diagnóstico diferencial de la debilidad muscular, entre síndrome paraneoplásico, síndrome de canal medular cervical estrecho, miositis autoinmune, por fármacos (intoxicación por estatinas) o síndrome de Guillain-Barré. Se realizaron colonoscopia total, radiografía torácica, ecografía abdominal y biopsia muscular, con resultados normales; se realizó endoscopia y la biopsia gástrica evidenció gastritis crónica atrófica folicular, metaplasia de tipo intestino delgado y Helicobacter pylori positivo. La EMG de miembros inferiores reportó signos de inestabilidad de membrana asociados a ondas y fibrilaciones positivas, compatible con miopatía generalizada activa. Las pruebas de VIH, VDRL, AntiDNA fueron negativos y anticuerpos antinucleares (ANA) fueron positivos: 1/1280 (1/40). La suma de los paraclínicos, la sintomatología y la EMG permitieron descartar otras causas de debilidad muscular y diagnosticar polimiositis, ya que no se presentaron manifestaciones cutáneas ni alteraciones que hicieran sospechar otro tipo de miopatía inflamatoria, por lo que se realizó manejo con prednisona oral 75 mg/día y se dio alta tras dos semanas con mejoría en la fuerza de las extremidades, CPK: 4944 U/L y prednisolona oral.
Dos semanas posteriores al egreso continuó con disminución de la fuerza en las cuatro extremidades y en los flexores del cuello, con Antígeno Ca 19.9 < 2 UI/mL (< 37 UI/mL), TSH 4.2 U/L, Tiroxina libre 1.24 UI/mL, CPK: 7758 U/L, por lo cual reumatología disminuyó prednisolona a 50 mg/día y adicionó azatioprina 50 mg/día, metotrexato 15 mg/semanal y ácido fólico 1 mg/día. A los dos meses se reportaron ANA, AntiDNA y Anti JO-1 negativos, Antígeno carcinoembrionario y alfa feto proteína normales, CPK: 6842 U/L y LDH: 780 U/L con fuerza 5/5 en todas las extremidades, por lo cual se disminuyó dosis de los medicamentos (Figura 1). Cuatro meses después, el paciente abandonó el tratamiento porque percibió recuperación completa de su fuerza y por desconocer que padecía una enfermedad crónica que necesitaba tratamiento permanente, además de inconvenientes administrativos con la aseguradora de salud que impidieron continuar los controles médicos.
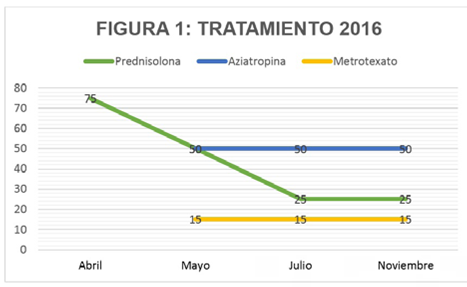
El paciente permaneció asintomático por 3 años y en junio de 2019 consultó por debilidad muscular en las cuatro extremidades de 8 días de evolución, pérdida de peso de 5 kg en 20 días, con paraclínicos de ingreso alterados, por lo cual, basándose en su antecedente médico, se ordenó prednisolona 1 mg/ kg/día para manejo de agudización de polimiositis por ausencia de tratamiento.
Buscando otra causa de debilidad muscular, se realiza ecografía de abdomen superior, radiografía torácica y biopsia muscular, con resultados normales; también se hacen pruebas de Hepatitis C, Antígeno de superficie de Hepatitis B, Prueba no treponémica PRP, VIH, Anticuerpos antimúsculo liso y anticuerpos antimitocondriales negativos; anticuerpos antiperoxidasa, anticuerpos antitiroglobulina, anticuerpos J0-1, perfil tiroideo y antígeno prostático normales.
Reumatología indicó pulsos de metilprednisolona (750 mg/día) durante 3 días y continuar con prednisolona oral. Tras 10 días de tratamiento intrahospitalario y mejoría se dio alta con CPK: 3615 U/L e hipertransaminasemia, con metotrexato oral (12,5 mg/semanal en dosis única) y ácido fólico (Tabla 2).
Tabla 2 Paraclínicos de ingreso y de seguimiento médico durante hospitalización.
| FECHA | CPK (UI/L) | TGP (UI/L) | TGO (UI/L) | PCR (md/dL) | Proteinuria (mg/dL) | Hematuria (uL) |
|---|---|---|---|---|---|---|
| 03-06-19 | 9543 | 330,4 | 357,8 | 2,64 | 25 | 25 |
| 06-06-19 | 7053 | 239 | 178 | |||
| 07-06-19 | 6813 | 248 | 190 | |||
| 10-06-19 | 5829 | |||||
| 13-06-19 | 3615 |
Fuente: autores.
15 días después del egreso, por aumento del CPK y disminución de la fuerza, medicina interna ordenó prednisolona oral 25 mg/día y aumentar metotrexato a 15 mg/semanal. Luego de una semana, por nuevo ascenso del CPK, se ordenó prednisolona 50 mg/ día por cinco días y posteriormente desescalar a 25 mg más tacrolimus. Un mes después, presenta Anticuerpos antimúsculo liso de 1/40 y por nuevo aumento de CPK se ordenaron pulsos de prednisolona oral: 75 mg/día por siete días, luego 50 mg/día otros siete días, continuar 25 mg/día, con resto de medicación igual (Figura 2).
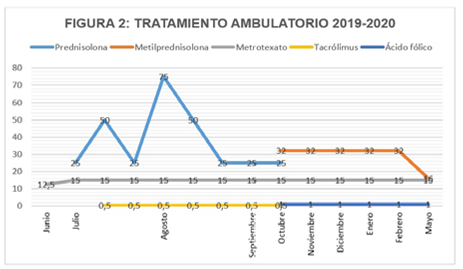
En octubre de 2019, la EMG de control reportó hallazgos compatibles con miopatía inflamatoria generalizada activa y por el lento descenso del CPK, reumatología cambió tratamiento a metilprednisolona oral 16 mg cada 12 horas, metotrexato 15 mg semanal y ácido fólico (Figura 2). Para valoración por hepatología ante sospecha de cirrosis biliar primaria por hipertransaminasemia persistente el paciente se realizó tomografía axial computarizada de abdomen, que reportó como hallazgo incidental un nódulo renal izquierdo que pudo corresponder a un cáncer de célula renal, por lo cual fue remitido a urología oncológica.
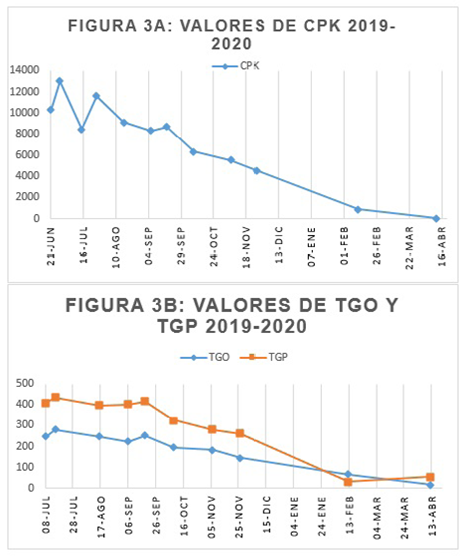
En febrero 2020 hubo descenso significativo del CPK, de TGP y valores normales de TGO; al mismo tiempo que el paciente se sometió a cirugía de resección de lesión renal quística simple benigna, descartando proceso neoplásico. Actualmente el paciente continúa en control con reumatología para disminución gradual de la dosis de corticoides, en abril del 2020 presentó CPK de valores normales por lo que se disminuyó metilprednisolona a 16 mg/ día, con persistencia de la fuerza muscular de 4/5 sin compromiso de otros sistemas ni complicaciones (Figura 3).
Discusión
La polimiositis es una miopatía inflamatoria autoinmune2. Un estudio europeo del año 2008 demostró que la proximidad a la latitud 0° y mayor exposición a radiación lumínica causaba dermatomiositis y que en los países más alejados la polimiositis era más frecuente, diferencias atribuidas a la radiación ultravioleta como etiopatogenia18, situación inversa en nuestro caso, ya que no presentó dermatomiositis a pesar de la cercanía a la línea del Ecuador.
Las manifestaciones musculares de la enfermedad afectan principalmente la cintura escapular y pélvica19. Como ocurrió en este caso, se presenta dificultad progresiva que evoluciona en semanas o meses para realizar tareas cotidianas como levantarse de la silla, subir escaleras o caminar sobre un piso irregular; además asociado a la debilidad pueden aparecer síntomas extramusculares1, de los cuales nuestro caso presentó hasta el momento de la elaboración de este artículo, únicamente pérdida de peso y artralgias.
Los criterios de Bohan y Peter no especifican cómo excluir otras formas de miopatía y ocasionalmente clasifican erróneamente a los pacientes12, pero clasificarían este caso como polimiositis probable. En los criterios del IMCCP los niveles de CPK, LDH y transaminasas son importantes, los pacientes se clasifican como caso si tienen una puntuación mínima de 5.512. Según la calculadora web de clasificación EULAR/ACR el paciente obtuvo puntuación de 7.620, por lo que se puede considerar como caso definitivo de polimiositis con una probabilidad de 75%13. Los criterios incluyen biopsia muscular, pero un resultado normal no descarta el diagnóstico, ya que en algunos pacientes pueden estar ausentes los cambios inflamatorios en el tejido muscular y esto puede ocurrir por el tratamiento con corticoides (incluso sin recibirlos). En estos casos lo más adecuado es realizar el diagnóstico de probable polimiositis y no de descartar21.
Según Findlay A. y colaboradores, se presenta elevación de la CPK de 5-50 veces en polimiositis activa, especialmente sin tratamiento17, lo que se asocia a una pérdida de la funcionalidad y posterior postración: situaciones que no presentó nuestro paciente, ya que podía realizar actividades cotidianas a pesar de la debilidad muscular y el aumento en la CPK, sin requerir asistencia mecánica ni apoyo para la movilización, por lo cual se infiere que los niveles de CPK no presentan relación marcada con un mayor compromiso muscular. Además de la elevación de CPK, existe una hipertransaminasemia que puede deberse a una lesión muscular22, ya que el descenso de las transaminasas se dio con la disminución de la CPK, aunque las primeras los hacían en menor medida. Dimachkie y Amato plantean que la elevación de la CPK apoya el diagnóstico con una sensibilidad de 69%, pero no es específica y su disminución se relaciona con una respuesta adecuada al tratamiento5, lo que se evidenció en nuestro caso ya que se modificaba el tratamiento dependiendo de este parámetro.
En la polimiositis, el riesgo relativo de malignidad es de 1,62 para hombres y hay riesgo elevado de linfoma no Hodgkin, neoplasias de pulmón y vejiga23. En nuestro caso, los marcadores tumorales evaluados fueron negativos, pero en 2019 una tomografía axial computarizada de abdomen reporta una masa sólida en el riñón izquierdo, resecada quirúrgicamente y diagnosticada como lesión quística benigna en un estudio de inmunohistoquímica, por lo que, hasta la fecha de realización del artículo, puede descartarse dicha asociación.
Dependiendo del compromiso, el tratamiento inicial consiste en la administración de dosis altas de esteroides buscando reducir la dosis a la cantidad más pequeña y efectiva para evitar los efectos adversos asociados al síndrome de Cushing exógeno cuando el tratamiento es prolongado, por lo tanto, cuando no hay mejoría después de 3 meses o hay recaída mientras se estrecha la medicación, se debe agregar un agente inmunomodulador (metrotexato, azatioprina, tacrolimus)17. Aunque otra alternativa es iniciar estos agentes desde el momento del diagnóstico junto con corticoides, considerando la gravedad del cuadro11. En nuestro paciente, el manejo inicial por parte de medicina interna fue el uso de corticoides y al no responder a la monoterapia, reumatología adicionó inmunomoduladores al tratamiento, clasificando el caso como enfermedad “refractaria definitiva”, término que se refiere a la incapacidad de remisión luego de 3 meses de terapia con esteroides17. Los objetivos del tratamiento son eliminar la inflamación, restaurar el rendimiento muscular, reducir la morbilidad y mejorar la calidad de vida3.
Los síntomas asociados pueden afectar la calidad de vida, ser atípicos o presentarse como síndrome paraneoplásico. En diferentes reportes de casos se mencionan disfagia de líquidos y sólidos, disfonía y odinofagia24,25, dificultad para mantener la cabeza erguida, disnea y cianosis26; limitación de movimientos orofaciales, para articular palabra y mantener tono postural27, sintomatología poco específica como cefalea, diarrea, vómito y tos, asociados a imposibilidad para la marcha y postración28y taquicardia de QRS estrecho e intervalo PR prolongado, como el caso reportado por Reyes Tovilla y colaboradores en 202029.
En la literatura también se reporta la presentación de síndromes paraneoplásicos como el caso de un varón de 76 años con CPK 17000 UI/L, con una masa en esófago distal correspondiente a un carcinoma epidermoide30; el de un varón de 63 años con tos y disfagia en quien se encontró un nódulo en el lóbulo superior del pulmón izquierdo, compatible con adenocarcinoma31; o el caso reportado en México de una mujer de 65 años con un nódulo en glándula mamaria derecha que correspondió a carcinoma ductal infiltrante, en quien la debilidad muscular mejoró luego de tres ciclos de quimioterapia32. En nuestro caso, a pesar de los niveles elevados de CPK, el abandono del tratamiento, la agudización, el quiste renal y el fracaso del tratamiento en distintos momentos, no se presentó sintomatología asociada incapacitante ni síndrome paraneoplásico, a diferencia de lo mencionado en la literatura.
Conclusiones
Se presentó el caso de una patología infrecuente donde se evidencia el desconocimiento de diversos aspectos de la enfermedad que, al no tener una historia natural homogénea, hace necesaria la individualización de su manejo inicial y seguimiento, más aún en hombres con un cuadro clínico atípico como este, al tener estudios de autoinmunidad sin hallazgos relevantes y con biopsia muscular normal, por lo cual es importante correlacionar las manifestaciones clínicas con las ayudas diagnósticas para descartar otras causas secundarias frecuentes de debilidad muscular y llegar al diagnóstico de polimiositis.
Aunque en este caso no hubo complicaciones ni compromiso de otros sistemas, la asociación de polimiositis con sintomatología incapacitante y síndrome paraneoplásico deja en evidencia la necesidad de disponer de pruebas de laboratorio especializadas, del tratamiento oportuno y seguimiento estricto para evitar las posibles complicaciones.

