











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
El síndrome de Sjögren (SSJ) es un trastorno crónico, autoinmunitario y multisistémico que afecta a las glándulas secretoras, en particular, a las glándulas lagrimales y salivales, produciendo xeroftalmia y xerostomía, respectivamente1. Sumado a la disfunción de las glándulas exocrinas, los pacientes pueden presentar una variedad de manifestaciones extraglandulares (sistémicas) como artralgias o artritis, fenómeno de Raynaud, linfadenopatías, afección pulmonar, vasculitis, afección renal, linfomas, esplenomegalia, neuropatía periférica y miositis. Todo esto debido a las lesiones autoinmunitarias de múltiples sistemas orgánicos como el sistema nervioso central, vascular, articular, muscular, cutáneo, pulmonar y renal1.
El SSJ puede presentarse como un trastorno primario (SSJ primario), caracterizado por ser de progresión lenta, acompañado por una infiltración linfocitaria; o puede estar asociado a otras enfermedades autoinmunitarias como la artritis reumatoide, el lupus eritematoso sistémico y la esclerodermia (SSJ secundario)1,2.
La primera descripción del Síndrome de Sjögren (SSJ) se dio en 1888, no obstante, fue hasta 1933 que Henrik Sjögren refirió la tríada de queratoconjuntivitis seca, xerostomía y artritis en 19 personas2,3. Hoy en día, este cuadro sigue teniendo gran utilidad clínica, sin embargo, a lo largo de las pasadas décadas se han debatido y revisado los criterios diagnósticos del SSJ, y actualmente los más usados son los criterios de clasificación de 2016 de American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) del síndrome de Sjögren (SSJ) primario2.
Etiológicamente, se ha reportado que la naturaleza autoinmune de la enfermedad en los pacientes se da por la presencia de anticuerpos como los antinucleares, los anti-Ro y anti-La, así como también la presencia del factor reumatoide, el anti-centrómero y el receptor anti-muscarínico4. Otra característica de la enfermedad que implica una etiología autoinmune es la presencia de un infiltrado linfocítico en los órganos comúnmente involucrados, las glándulas salivales y lagrimales5.
Existe una gran variedad en la incidencia de SSJ entre varios estudios. Las tasas de prevalencia en Colombia varían, por ejemplo, un estudio realizado por Fernández y cols. reportó prevalencia que va desde 0,12 % hasta 0,5 %, según el género, grupo etario y departamento de residencia6. Una revisión sistemática realizada por Qin y cols.7 informó una razón de incidencia de 6,92 (IC 95 % 4,98 a 8,86) por 100 000 personas al año. Esta afección ocurre con preferencia en el sexo femenino entre el 9095 %8. Un estudio transversal realizado en Colombia durante los años 2012 a 2016, en el que se buscaba la prevalencia de poliautoinmunidad en pacientes con SSJ, tomó los datos del Sistema Integral de Información de la Protección Social del Ministerio de Salud, donde se identificaron 58 680 casos de SSJ, la mayor cantidad de estos se registraron en Bogotá D. C. (24 885), Antioquia (9040) y Valle del Cauca (5277), y presentaron una prevalencia nacional del 0,12 %, y fue más prevalente en el grupo etario de los 65 a 69 años6.
Hay poca disponibilidad de literatura reciente que recopile los aspectos más importantes acerca del SSJ. El objetivo de este manuscrito es realizar una revisión de la literatura sobre los aspectos generales del síndrome de Sjögren.
Metodologia de búsqueda
Se realizó una búsqueda de la literatura en las bases de datos Pubmed/Medline, Science Direct, Scopus, Embase, Redalyc y Scielo, entre el 15 de enero y el 15 de marzo del 2020; se hizo una búsqueda de los términos: síndrome de Sjögren, glándulas salivales, queratoconjuntivitis seca, autoinmunidad.
Se incluyeron estudios primarios y secundarios tales como revisiones de tema, revisiones sistemáticas, metaanálisis, ensayos clínicos, estudios de cohorte, casos y controles y estudios transversales. De igual manera, se tuvieron en cuenta estudios en humanos, mayores de 18 años, publicados en los últimos 10 años y en idioma español e inglés. Por otro lado, se excluyeron documentos como reportes de caso, cartas al editor, cartas científicas y aquellos artículos que evaluaran patologías con sintomatología similar al SSJ, pero de diagnóstico distinto (síndrome Sjögren-Larsson, xerostomía por amiloidosis, sarcoidosis, fibrosis quística, parotiditis).
De acuerdo con la estrategia de búsqueda, inicialmente, se obtuvo un total de 1782 artículos. Posteriormente, tras la aplicación de los criterios de inclusión, se obtuvo un total de 120 artículos. Tras la aplicación de los criterios de exclusión (n: 61), quedaron 59 artículos. Finalmente, se retiraron 30 artículos por estar repetidos, por lo que se utilizaron 29 artículos para la actual revisión (ver figura 1).
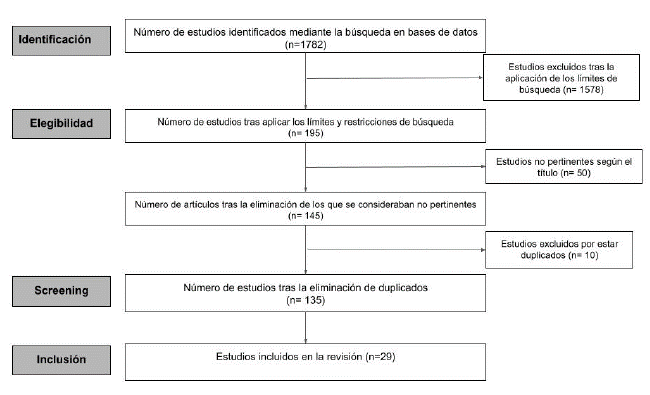
Desarrollo del tema
Factores de riesgo
Distintos factores se han encontrado asociados con esta enfermedad, los cuales se muestran en la tabla 1. 2,6,7,9-11
Tabla 1 Factores de riesgo2,6,7,9-11.
| Factores | Descripción |
|---|---|
| Genéticos | Agregación familiar: antecedente familiar de primer grado; genes HLA; genes no HLA: CHMR3, EBF1, FAM167A-BLK, TNFSF4, MECP2. |
| Medioambientales | Virus: EBV, virus de la hepatitis C, citomegalovirus, retrovirus, HTLV-1. |
| Hormonales | Disminución de estrógenos |
*CHMR3: receptor del gen muscarínico M3, EBF1: factor primitivo de célula B, FAM167A-BLK: familia con secuencia familiar miembro 167 de tirosin quinasa A-B linfoide, TNFSF4: superfamilia del factor de necrosis tumoral; MBL: lectina ligadora de manosa; MECP2: proteína 2 unida a Metil-CpG; EBV: virus de Epstein Barr; HTLV-1: virus linfotrópico humano T.
Fuente: autores.
Las alteraciones genéticas en las regiones HLA clase I, y III se asocian con producción de autoanticuerpos de la enfermedad, sin embargo, existen otras alteraciones presentes en diferentes loci de genes que codifican moléculas involucradas en la señalización de IFN tipo I y tipo II y procesos diana (como IRF5, IL12A y STAT4). Asimismo, la señalización de NF-kB, el tráfico de linfocitos (como CXCR5) y la activación y diferenciación de células productoras de anticuerpos (como BLK) están sustancialmente asociados al desarrollo de SSJ3.
Los factores medioambientales pueden estimular la expresión de los genes del interferón de los tipos I y II, posiblemente relacionados con infecciones víricas, que además pueden ser prolongadas por inmunocomplejos como anti-Ro y anti-La unidos a hYRNA (hY: human y cytoplasmic)2,11.
Por otro lado, se han encontrado factores hormonales relacionados con esta patología, por ejemplo, el hecho de que las glándulas salivales tienen un receptor de estrógeno, y en las mujeres posmenopáusicas se produce una deficiencia de estrógenos, que induce la sobreexpresión del factor de transcripción proteína 48 (RbAP48) que genera apoptosis de células epiteliales, infiltrados linfocitarios en glándulas exocrinas y favorece la formación de autoanticuerpos11.
Fisiopatologia y etiopatogenia
La fisiopatología es compleja y no se ha comprendido por completo, sin embargo, en la actualidad se considera que las células T y las células B desempeñan un papel importante en la fisiopatología de esta enfermedad12.
Células T en el sindrome de Sjögren
La presencia, y a veces el predominio, de células T CD4 + en los infiltrados de glándulas salivales enfatiza su contribución potencial a la patogénesis del SSJ. Un metaanálisis mostró la asociación entre SSJ y varios alelos principales del complejo de histocompatibilidad clase 2 (MHC2), lo que sugiere que la presentación de autoantígenos es importante en la patogénesis del SSJ13. Se ha planteado que las células Th1 son el subtipo principal que contribuye a la patogénesis, ya que se unen a las moléculas de MHC2 que inician una respuesta inmune14. Asimismo, las células T reguladoras (Treg) se han identificado en las glándulas salivales de pacientes con SSJ y la mayor presencia de estas células se ha asociado con un mayor grado de inflamación en las lesiones locales15. Se sabe que las T reguladoras tienen efectos supresores sobre la proliferación y la función de las células T efectoras. Se ha informado que el número de Tregs circulantes aumenta, mientras que su función no parece estar afectada en SSJ, lo que sugiere que los Tregs no juegan un papel importante en la patogénesis del SSJ14.
Células B en el sindrome de Sjögren
Son células inmunes adaptativas responsables de la secreción de anticuerpos y la presentación del antígeno. El factor de activación de células B (BAFF) es una citocina que promueve la proliferación, maduración y supervivencia de las células B, y es inducida principalmente por los interferones tipo I y tipo II14. Estos interferones son producidos por células dendríticas plasmacitoides14,15. Se ha sugerido que ciertos virus (por ejemplo, Epstein-Barr) y la formación de complejos inmunes activan los receptores Toll-like (como los TLR 3, 7 y 9), lo que lleva a la activación de la inmunidad innata y la producción de interferon14,16 (ver figura 2).
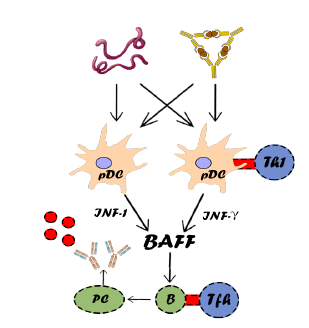
*El esquema muestra una visión general simplificada de la patogénesis de SSP (síndrome de Sjögren primario). Una causa desconocida (se sugiere que es un virus o un complejo inmunitario) puede conducir a la activación de los pDC, lo que aumenta los niveles de interferones. La producción de BAFF inducida por interferón conduce a una mayor proliferación y diferenciación de células B con la producción de autoanticuerpos como resultado. Abreviaturas: pDC: Células Dendríticas plasmacitoides; Th1: célula T-ayudante 1; IFN: Interferon; B: célula B; PC: Células Plasmáticas; Tfh: célula folicular T-helper.
Fuente: tomado de Both T, Dalm VASH, van Hagen PM, van Daele PLA14.
Figura 2 Visión general simplificada de la patogénesis de SSP (síndrome de Sjögren primario).
En pacientes con SSJ, el 55 % tiene una actividad aumentada de IFN tipo I versus el 4,5 % en controles sanos17. La presencia de esta llamada 'firma IFN tipo I' en monocitos de pacientes con SSJ se asoció con un mayor índice de actividad de la enfermedad del SSJ, la presencia de marcadores biológicos de actividad (niveles aumentados de IgG y/o hipocomplemento) y mayores niveles de ARNm de BAFF en monocitos17. Los niveles circulantes y de tejido glandular salival de BAFF están significativamente elevados en pacientes con SSJ, lo que se asocia con una mayor actividad de la enfermedad, pero también con un mayor riesgo de desarrollo de linfoma de células B14. Estos hallazgos respaldan la hipótesis de que la activación del sistema inmune innato conduce a una respuesta autoinmune por parte del sistema inmune adaptativo. Dado que BAFF es uno de los vínculos entre las respuestas inmunes innatas y adaptativas, podría ser un objetivo potencial para la terapia en SSJ18 (ver figura 2).
Tras el reconocimiento del antígeno en el centro germinal, las células B proliferan y se diferencian en una célula B específica para este antígeno14. La formación de centros germinales es probablemente importante en la patogénesis de SSJ debido a la promoción de la estimulación crónica y la activación (por las células T auxiliares foliculares) de las células B, lo que permite que estos pacientes a menudo presenten altos niveles en suero de IgA y/o IgG14. La hiperglobulinemia puede conducir a la formación de complejos inmunes con tendencia a precipitarse en los órganos principales y provocar daños irreversibles14. Además, la presencia de autoanticuerpos (anti-Ro52, anti-Ro60 y anti-La) se incluye en los criterios de diagnóstico para SSJ14. La presencia de estos autoanticuerpos se asocia con enfermedad de inicio temprano, agrandamiento de la glándula parótida, manifestaciones extraglandulares e infiltración glandular linfocítica15.
Cuadro clinico
La presentación clínica varía desde síntomas leves, como la xeroftalmía, xerostomía, queratoconjuntivitis, hasta síntomas sistémicos graves que afectan múltiples sistemas orgánicos como vasculitis, glomerulonefritis y una serie de manifestaciones neurológicas19. Los síntomas varían dependiendo de la clasificación del SSJ; si es un SSJ primario, se caracteriza principalmente por la xeroftalmía y la xerostomía20. Por otro lado, si es un SSJ secundario, puede haber xeroftalmía y/o xerostomía, generalmente menos intensa que en el SSJ primario, asociada a una enfermedad o situación autoinmune bien identificada20.
La mayoría de los pacientes con SSP suelen presentar, en primer lugar, xerostomía y/o xeroftalmia21. Sin embargo, algunos pacientes, especialmente niños y adultos jóvenes, pueden presentar fiebre continua y bien tolerada, junto con sudores nocturnos, fatiga, malestar y pérdida de peso. En estos pacientes, los síntomas sistémicos son nulos o insignificantes, lo que a menudo provoca un diagnóstico tardío de SSJ22.
Es importante tener en cuenta que la xeroftalmía es provocada por la infiltración de linfocitos en la zona de las glándulas lagrimales, así como la destrucción de acinos y conductos de estas. Las manifestaciones clínicas incluyen fotofobia, sensación de arena en los ojos, prurito, quemazón y dolor al parpadeo; en el examen ocular se evidencia hiperemia conjuntival y disminución de la agudeza visual20. La xerostomía se da por la disminución de la secreción de las glándulas salivales, que, al ser lesionadas por el infiltrado linfocitario, producen manifestaciones clínicas dadas por la sensación de sed, dificultad con la masticación y la deglución, para lo cual el paciente tiene necesidad de ingerir líquidos constantemente20.
Existe una variedad de características extraglandulares (o sistémicas) que se pueden desarrollar durante la presentación de la patología; cuando las características sistémicas aparecen antes de la xerostomía o cuando los síntomas leves pasan desapercibidos por el paciente, el médico o ambos, se puede hablar de un SSJ "oculto", a diferencia de la presentación usual donde se manifiesta primero la xerostomía21. El compromiso sistémico específico del SSJ puede incluir sistema vascular, articular, muscular, cutáneo, pulmonar, renal y el sistema nervioso central y periférico (ver tabla 2)22,23.
Tabla 2 Características extraglandulares22,25.
| Sistema | Patología |
|---|---|
| Sistema nervioso central | Enfermedad focal y difusa |
| Vascular | Fenómeno de Raynaud-hipertensión pulmonar |
| Articular | Artritis-artralgias |
| Muscular | Fibromialgiamiositis |
| Cutáneo | Vasculitis-eritema anular |
| Pulmonar | Enfermedad pulmonar intersticial |
| Renal | Nefritis tubulointersticial |
| Nervios periféricos | Polineuropatía |
Fuente: autores
Diagnóstico
Este síndrome generalmente se diagnostica por medio de pruebas serológicas y por la clínica, con base en distintos criterios. Según los criterios de clasificación propuestos por el grupo de Consenso Americano y Europeo, publicados en el año 2012 (ver tabla 3), si se cumplen 3 criterios, se considera SSJ probable; por debajo de este valor se descarta el diagnóstico, pero es necesario cumplir con 4-6 criterios, incluidos el 5 o el 6, para diagnosticar SSJ con una sensibilidad del 93,5 % y una especificidad del 94 %24,25. También se encuentran los criterios de clasificación de la Universidad Americana de Reumatología / Liga Europea Contra el Reumatismo, del 2016 (ver tabla 4), con una sensibilidad del 96 % y una especificidad del 95 %; un resultado ≥4 puntos sugiere SSJ primario26. Ambos criterios representan una buena alternativa de diagnóstico por la alta sensibilidad y especificidad, sin embargo, los criterios de la Universidad Americana de Reumatología / Liga Europea Contra el Reumatismo son más sensibles.
Tabla 3 Criterios de clasificación del grupo de Consenso Americano-Europeo 2012
| 1. Síntomas orales (1 positivo): sensación de boca seca por un periodo superior a 3 meses, parotidomegalia recurrente, necesidad de ingesta de líquidos constante. |
| 2. Síntomas oculares (1 positivo): ojos secos durante más de 3 meses, sensación de arenilla ocular recurrente, necesidad de utilizar lágrimas artificiales más de 3 veces al día. |
| 3. Signos oculares (1 positivo): prueba de Schirmer menor o igual a 5 mm a los 5 minutos. Puntuación mayor o igual a 4 en tinción de rosa de Bengala (escala de Bisterveld). |
| 4. Alteración de glándulas salivales (1 positiva): gammagrafía parotídea con déficit difuso de captación (grado III: marcado enlentecimiento con disminución tanto de concentración como de excreción del trazador; grado IV: ausencia de actividad glandular), sialografía con alteración difusa ductal y flujo salival sin estimular de 1,5 ml o menos en 15 minutos. |
| 5. Histopatología: biopsia salival grados III-IV (clasificación Chisholm y Mason): sialoadenitis linfocítica crónica. |
| 6. Inmunología (1 positiva): anti-Ro, anti-La |
Fuente: tomado y adaptado de Ruiz Gutiérrez y cols24.
Tabla 4 Criterios de clasificación del síndrome de Sjögren primario según ACR/EULAR, 2016
| Criterios | Puntuación |
|---|---|
| Focos de inflamación con infiltración linfocítica en la glándula salival labial y ≥1 foco en 4 mm2 | 3 |
| Presencia de anticuerpos anti-Ro positivos | 3 |
| Tinción conjuntival y corneal ≥5 en escala de Whitcher y cols. o ≥4 en escala de van Bijsterveld, por lo menos en un ojo | 1 |
| Prueba de Schirmer ≤5 mm pasados 5 min, por lo menos en un ojo | 1 |
| Secreción salival no estimulada valorada con el método de Navazesh y Kumar ≤0,1 ml/min | 1 |
| Casos excluyentes: irradiación anterior de cabeza y cuello, infección activa por VHC (confirmada mediante PCR), SIDA, sarcoidosis, amiloidosis, reacción de injerto contra huésped, enfermedad sistémica asociada a IgG4. | |
Fuente: tomado de Le Goff y cols26.
Entre los exámenes paraclínicos solicitados se incluye la sialometría, sialografía parotídea, gammagrafía parotídea y biopsia de glándula salivar menor. Por otro lado, dentro de los exámenes de laboratorio que tienen mayor valor diagnóstico se incluye los anticuerpos antinucleares (ANA), anti-Ro, anti-La, los cuales son positivos en un 50-70 % de los pacientes, con una sensibilidad baja (33-46 %), pero con una especificidad del 100 %24; también es frecuente solicitar la hipergammaglobulinemia policlonal27.
Tratamiento
El manejo de las enfermedades autoinmunes está dirigido, principalmente, a modificar el curso de estas, controlar laactividad inflamatoria, frenar laprogresión y evitar las secuelas que se puedan presentar. Sin embargo, hasta la fecha, los tratamientos empleados en el SSJ son empíricos y enfocados a tratar los síntomas, debido a que actualmente no hay ninguno que haya demostrado ser efectivo en la modificación del curso de la enfermedad26. A continuación, se presentan las estrategias de tratamiento disponibles en la actualidad para el manejo del SSJ.
Tratamiento para la afección ocular
La primera línea de tratamiento de la xeroftalmia está constituida por lágrimas artificiales, cuya principal función es lubricar el ojo. Las más usadas en la práctica clínica son las que contienen celulosa y hialuronato sódico, y se deben evitar las lágrimas con conservantes para evitar la irritación local. En casos refractarios se pueden utilizar colirios de suero autólogo del paciente24. Otros tratamientos disponibles incluyen el uso de ciclosporina al 0,05 % aplicando una gota en cada ojo dos veces al día24.
Los secretagogos son otra alternativa para el tratamiento de la afección ocular, el clorhidrato de pilocarpina es un agonista de los receptores muscarínicos de las glándulas exocrinas con funciones parasimpaticomiméticas. Se utiliza en dosis de 5 mg cada 6 horas (con agua, antes de las comidas), y se puede incrementar la dosis cada 4-8 semanas hasta un máximo de 30 mg por día repartidos en 4 tomas. Puede producir efectos secundarios colinérgicos como náuseas, cefalea, sudoración, mareo y poliuria que llevan a muchos pacientes a abandonar el tratamiento24. Finalmente, se puede cerrar el conducto lagrimal, ocluir los canalículos lagrimales de forma temporal mediante tapones (de colágeno o silicona) o de forma permanente por medios quirúrgicos (cauterización térmica o ligadura con sutura)24.
Tratamiento de la afectación bucal
Algunas de las recomendaciones para tratar la xerostomía incluyen tomar alimentos ácidos no azucarados que incrementen la secreción salival, mantener una higiene oral rigurosa, evitar sustancias como el tabaco, el café y las bebidas alcohólicas. Además, se pueden emplear secretagogos, como en el caso de la xeroftalmia24.
Tratamiento de las manifestaciones extraglandulares
Las manifestaciones extraglandulares o sistémicas, al igual que las glandulares, representan una queja importante en los pacientes que las presentan, por tanto, deben ser tratadas. Los siguientes fármacos pueden ser útiles para dichas afecciones: los corticoides, por ejemplo, pueden ser necesarios como tratamiento en pacientes que presenten afectación pulmonar, renal o en casos de vasculitis; la hidroxicloroquina, por sus efectos a nivel inmunológico, puede ser utilizada en la afectación articular del SSJ24. El metotrexato suele ser empleado con frecuencia en pacientes con SSJ y artritis, sin embargo, existe escasa evidencia que respalde su utilización24. Los fármacos biológicos como los anti-TNF, el infliximab y el etanercept no han demostrado ser eficaces en dos ensayos clínicos controlados y aleatorizados realizados en pacientes con SSJ28,29. El uso del rituximab en estos pacientes ha resultado controvertido, sin embargo, en cuanto al belimumab, anticuerpo monoclonal contra BAFF, se han mostrado resultados prometedores en un estudio reciente realizado en pacientes con manifestaciones sistémicas, hipertrofia de glándulas salivales, duración de la enfermedad de menos de 5 años o biomarcadores de activación de células B24.
Complicaciones
La xerostomía puede generar a largo plazo candidiasis oral, pérdida dental y enfermedad periodontal; además, la xeroftalmia, si no se trata, puede provocar úlceras corneales y perforaciones24. Los síntomas extraglandulares graves sin tratamiento temprano pueden desembocar en fracaso orgánico que puede incluir: falla renal crónica, fibrosis pulmonar, enfermedad neurológica progresiva e incluso llevar al paciente a la muerte. Las neoplasias hematológicas se han descrito como la principal complicación del SSJ, asociadas a un riesgo 10-50 veces más alto que en las personas sanas24.
El riesgo de desarrollar linfoma o enfermedades linfoproliferativas en el transcurso del SSJ se valoró en distintos estudios, de los cuales se concluyó que es de aproximadamente el 5 % en los primeros 5 años, 10 % a los 15 años y 18 % a los 20 años del diagnóstico. Su localización más frecuente es extraganglionar, en glándulas salivales, tracto gastrointestinal, pulmón, riñón u órbita. Aquellos pacientes que presentan linfoma están asociados a una menor supervivencia; los factores de riesgo independientes de mortalidad son los linfomas de medio/alto grado, la presencia de síntomas B (fiebre, sudoración nocturna y pérdida de peso), así como un diámetro tumoral mayor de 7 cm. Algunos de los factores que han sido mencionados como favorecedores para el desarrollo de linfoma son la inflamación glandular recurrente, púrpura palpable, presencia de linfadenopatías, crioglobulinemia, hipocomplementemia, linfopenia, positividad para anticuerpos (anti-Ro- y anti-La) y positividad para factor reumatoide24.
Pronóstico
La sintomatología en el SSJ se debe a la hipofunción y/o atrofia glandular. En general, el pronóstico es benigno, salvo en los casos en los que existe afectación visceral (SSJ secundario); en estos casos, el pronóstico estará en relación con la enfermedad asociada, por ello la evolución clínica no es predecible, aunque la mayoría suelen seguir un curso estable de varios años de evolución con los mismos síntomas23.
Conclusiones
El SSJ tiene una importante prevalencia entre las enfermedades autoinmunes más comunes, afecta principalmente a personas del género femenino mayores de 50 años, por lo cual se considera que hay una influencia hormonal importante en el proceso fisiopatológico. El cuadro clínico, a pesar de ser inespecífico, está dado principalmente por xerostomía y xeroftalmia, debido al compromiso de las glándulas secretoras, en particular, de las glándulas salivales y lagrimales. En cuanto a los criterios diagnósticos, se encontró que los de la Universidad Americana de Reumatología y la Liga Europea Contra el Reumatismo (2016) tienen una sensibilidad y especificidad que superan a los criterios propuestos en el consenso americano y europeo. Respecto al manejo, está enfocado principalmente al a controlar los síntomas, frenar la progresión y disminuir las secuelas que se pueden presentar, ya que, aún no se ha establecido un tratamiento específico para frenar el curso de la enfermedad. Las complicaciones de esta enfermedad se deben identificar, ya que, influyen directamente en el pronóstico del síndrome.

