











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El síndrome de Down (SD), también denominado trisomía 21, constituye la causa más frecuente de discapacidad cognitiva de origen cromosómico. La incidencia promedio es de 1 por cada 800 nacidos vivos a nivel mundial, aunque puede variar entre 1/700 a 1/1500 según características como el diagnóstico prenatal y la edad de concepción1,2. Se ha descrito que una edad materna/paterna mayor de 35 años presenta mayor asociación, así como una paridad materna mayor de cinco hijos3. En Colombia, según datos de 2020 del Instituto Nacional de Salud, el SD presenta una incidencia de 6,03 a 7,86 por cada 10 000 nacidos vivos4,5. Por otra parte, datos de 2012 de la Universidad Javeriana, describen una incidencia de 1 en 700 nacidos vivos y un estudio realizado entre 1991 a 1995 en Cali determinó una incidencia de 1 por cada 650 nacido vivos6,7.
En cuanto a su etiología, el SD es una cromosopatía producida por trisomía del cromosoma 21. En la mayoría de los casos por no disyunción meiótica y el porcentaje restante es secundario a otros mecanismos descritos en la tabla 1 8.
Tabla 1 Etiología del síndrome de Down
| Característica cromosómica | Porcentaje de casos (%) | Descripción |
|---|---|---|
| No disyunción meiótica | 96 | El riesgo aumenta con la edad materna. |
| Translocación | 3-4 | Usualmente ocurre entre cromosoma 21 y otro cromosoma que normalmente es el 14 o el 22. |
| Mosaicismo | 1-2 | Porcentaje de células afectadas es variable. |
| Trisomía parcial | <1 | Duplicación de un segmento delimitado del cromosoma 21. |
Tomado y modificado de: Down Syndrome. N Engl J Med 2020;382:2344-52
En el cromosoma 21 se encuentran más de 200 a 300 genes, cuyas alteraciones explican el fenotipo variable de la enfermedad 9. Se han propuesto diversos genes dosis-sensibles causantes de las complicaciones en SD, por ejemplo, la afección motora está relacionada con la sobreexpresión de los genes ITSNi (21q22.1), SYNJi (21q22.2) y DSCRi (21q22.12). Por su parte, la sobreexpresión del gen Ets2 (21q22.2) demuestra una asociación con las alteraciones esqueléticas y los estudios a la fecha refieren que los genes RUNX1 (21q22.3), ERG (21q22.2) y ETS2 (21q22.3) estarían implicados en la susceptibilidad a patología hematológica 7,9.
Las fascies de los pacientes se caracterizan por microcefalia leve, braquicefalia, cuello corto, ojos almendrados, fisuras palpebrales hacia arriba y afuera, microstomía, orejas con hélix muy plegado y con conducto auditivo externo estrecho, manos pequeñas, falanges cortas y clinodactilia. En el caso del neonato, los criterios de Hall (ver tabla 2) han demostrado ser altamente prevalentes, y la presencia de 4 o más criterios se considera muy sugestivo de la enfermedad 10. Ante la sospecha clínica, el diagnóstico se confirma con base en el análisis cromosómico a través de cariotipo 11.
Tabla 2 Criterios de Hall: criterios clínicos para el diagnóstico neonatal del SD
Fuente: Hall B. Mongolism in Newborn Infants: An Examination of the Criteria for Recognition and Some Speculations on the Pathogenic Activity of the Chromosomal Abnormality. Clin Pediatr (Phila). 1966;5(1):4-12.
Con los años, la calidad y esperanza de vida en las personas con SD ha cambiado de forma favorable logrando una mayor autonomía; específicamente la esperanza de vida ha aumentado considerablemente de 4 años en 1950 a 58 años en 2010 10,11. Además, hay mayor conocimiento de los riesgos y patologías asociadas, por ello las acciones preventivas permiten corregir e incluso evitar el problema de salud. Dentro de las condiciones médicas asociadas al síndrome se destacan la cardiopatía congénita, desordenes en el neurodesarrollo, déficit auditivo, complicaciones oncohematológicas, patologías autoinmunes y enfermedades endocrinológicas 12,13.
El compromiso neurológico constituye la principal causa de discapacidad en este grupo de pacientes. La adquisición de hitos de área motora es tardía y el promedio de coeficiente intelectual esta entre 35 a 70 puntos. Además, el desarrollo de Alzheimer es más frecuente con presentación a una edad más temprana8,11.
Una causa frecuente de mortalidad en los pacientes con SD son las complicaciones hematológicas y cardíacas. En primer lugar, el riesgo de padecer leucemia linfoide y/o mieloide aguda es muy alto. A partir de datos del registro nacional de Dinamarca se identificaron 2814 individuos con SD, de los cuales 60 desarrollaron cáncer y el desarrollo de leucemia presentó una razón de incidencia estandarizada de 17,6 (IC 12,4-24,4) 14. Por otra parte, hasta 50% de los pacientes con SD presentan una cardiopatía congénita, siendo más frecuente el canal atrioventricular seguido de comunicación interventricular, comunicación interatrial y tetralogía de Fallot 11.
Dentro de las complicaciones endocrinas, el hipocrecimiento constituye la más frecuente e incluso se considera una característica usual del paciente con SD. Por su parte, la patología tiroidea puede estar presente hasta en el 66% de los casos, siendo más frecuente el hipotiroidismo primario autoinmune. Adicionalmente, se estima que un 16,9% y un 28,2% de los niños con SD presenta obesidad o sobrepeso, respectivamente 15,16. Este grupo de complicaciones son determinantes y relevantes en la calidad de vida del paciente, lograr su prevención y proporcionar un tratamiento oportuno permite evitar complicaciones más graves. Es por ello necesario que el personal en salud, principalmente los profesionales en medicina general tengan conocimiento claro sobre estas complicaciones endocrinológicas en los pacientes pediátricos con SD.
En concordancia con lo descrito previamente, el objetivo de este artículo es revisar las complicaciones endocrinológicas prevalentes en el paciente pediátrico con síndrome de Down, relacionadas con el hipocrecimiento, desarrollo puberal, patología tiroidea, diabetes mellitus y obesidad; así como describir su seguimiento y tratamiento adecuado.
Metodología de búsqueda
Se realizó una búsqueda en la literatura científica desde agosto de 2020 hasta diciembre de 2021, en la base de datos PubMed y el buscador académico Google Scholar con los términos MeSH "Down syndrome", "Endocrine System Diseases", "Thyroid Gland", "Diabetes Mellitus", "Puberty". Se seleccionaron artículos de revisión, guías de práctica clínica, reportes de casos o estudios originales de los últimos 5 años. Se eliminaron referencias duplicadas o escritos en un idioma diferente a inglés/español. Cada una de las autoras realizó la lectura inicial del título y el resumen de los documentos, esto con el fin de determinar si eran de interés para el estudio, y en caso de que las dos autoras considerarán que no presentaba información relevante, por ser duplicada, el artículo era excluido. Adicionalmente se excluyeron aquellos artículos con contenido referente a patología en adultos. Finalmente se escogieron 44 artículos cuyo tema central fuera la asociación de patología endocrinológica en pacientes pediátricos con SD. Ver figura 1.
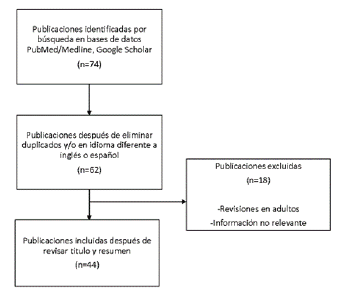
Desarrollo de tema
En los pacientes pediátricos con SD se describe una mayor prevalencia de patología endocrina: un 20,9% en SD comprado con 5% en la población general 13,16. Las más frecuentes constituyen alteraciones en el crecimiento, obesidad, patología tiroidea y un mayor riesgo de desarrollar diabetes mellitus (ver figura 2). En consecuencia, se requiere una adecuada intervención en este grupo de pacientes, la cual permita establecer un diagnóstico y tratamiento oportuno 12.
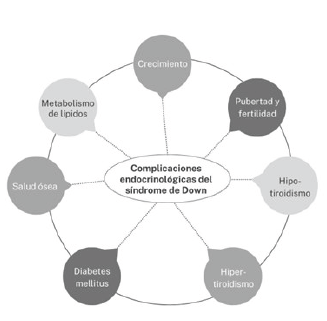
Crecimiento
Los pacientes con SD presentan una tasa de crecimiento diferente al resto de la población 17. Se caracterizan por presentar un retraso en la maduración esquelética e hipocrecimiento de origen prenatal con una desviación estándar (DE) de talla/ edad entre -1,3 a - 4,9 asociado a microcefalia. El hipocrecimiento afecta a todos los grupos de edad, pero la mayor afección de la velocidad de crecimiento se presenta entre los 6 meses y 2 años 18. Posteriormente, el estirón puberal es más temprano y menos significativo, con pico promedio de velocidad de crecimiento de 8,5 cm/año en niños y de 7,3 cm/ año en niñas 18. Como consecuencia se presenta una talla adulta final baja, con una media de 163,4 cm en hombres y 151,8 cm en mujeres 18,19.
La causa del retraso del crecimiento no está conocida en su totalidad, pero se han descrito varios factores como la disfunción hipotalámica que genera una deficiencia neurosecretora de hormona de crecimiento (GH) 20,21 Esto demostrado por una falta de respuesta en estudios clínicos de estímulo con levodopa y clonidina, pero adecuada con el uso de hormona liberadora de hormona de crecimiento (GHRH) 21. Adicionalmente, estudios demuestran una disminución en el número de neuronas en el núcleo arcuato y ventromedial del hipotálamo, regiones responsables en la producción de GHRH 22. Además, los niveles de factor similar a la insulina tipo 1 (IGF-1) suelen estar persistentemente bajos en los pacientes con SD, al parecer relacionado con una alteración en la maduración del eje, lo que también afectaría el desarrollo cerebral y explicaría la microcefalia 21.
La presencia de otras comorbilidades como cardiopatía, apnea obstructiva del sueño, patología tiroidea y nutrición inadecuada; son problemas frecuentes en los pacientes con SD con un impacto negativo en el crecimiento. La deficiencia de zinc se ha descrito como otro factor asociado a talla baja en SD. Incluso, algunos autores describen como la suplencia de zinc por 6 a 9 meses en niños con deficiencia, mejora el apetito y la velocidad de crecimiento hasta en un 68%, la cual se asocia a mejoría en los niveles de IGF-1 19.
Dada las características del crecimiento en este grupo de pacientes, se cuenta con curvas de crecimiento específicas. Son varias las gráficas publicadas de diversas poblaciones (Estado Unidos, Reino Unido, Italia, Holanda, Suecia, Portugal, Japón, España) 23. Sin embargo, se estima que en general el hipocrecimiento es similar entre los pacientes con SD y no hay variaciones significativas entre los estudios realizados en diferentes países 23-25.
Una de las curvas más ampliamente usadas y recomendadas por la Academia Americana de Pediatría (AAP) son las realizadas por el Centro para Prevención y Control de Enfermedades (CDC) en 1998, con actualización en 2015. Con estos últimos datos se identificó mejoría en el peso en aquellos menores de 36 meses junto con una modesta mejoría en talla al nacer y posterior a los 5 años 25.
En la actualidad el uso de hormona de crecimiento no está indicado en el tratamiento estándar del paciente con SD 19. Aun así, algunos estudios han evaluado la respuesta en SD a esta terapia 20. Torrado y colaboradores, trataron 13 pacientes con SD y diagnóstico de déficit de GH por test dinámicos. Los hallazgos demostraron un incremento en la velocidad de crecimiento posterior a un año de tratamiento (5,4 ± 4,6 cm/año a 12,2 ± 3,2 cm/año) y aumento de perímetro cefálico (-3,1±1,3 DE a -2,3±4,2 DE). Durante el seguimiento no se registraron efectos adversos, la glucosa sérica y hemoglobina glicosilada no identificaron cambios significativos, pero no fueron evaluados efectos a largo plazo 20. Hallazgos similares en cuanto la velocidad de crecimiento fueron descritos por Annerén y colaboradores con 15 pacientes tratados por 3 años con somatropina, con mejoría de talla de -1,8 a -0,8 DE durante el tratamiento, pero sin efectos positivos en perímetro cefálico o desarrollo motor 21. Además, durante el manejo se logran niveles de IGF-1 normales, pero posterior a suspensión hay desaceleración del crecimiento 21.
Obesidad
Característicamente los niños con SD suelen presentar una distribución de masa corporal diferencial con mayor prevalencia de obesidad. Si bien es frecuente que durante los primeros dos años presenten un bajo peso, posteriormente se ha descrito un aumento progresivo del índice de masa corporal (IMC). Se estima que el 20% de los niños con SD y más del 50% de los adultos con SD tienen obesidad 16,26. A la fecha no se reconoce al SD como una causa sindrómica de obesidad, sino que se considera que su origen es multifactorial asociado a un dieta inapropiada y a un menor gasto calórico 26.
El IMC es la herramienta para tamizar el exceso de adiposidad y riesgo cardiometabólico en niños 27. En la actualización de curvas de SD realizadas en 2015, se realizaron por primera vez gráficas para IMC en niños con SD entre 2 y 20 años 25. Sin embargo, su exactitud para detectar sobrepeso u obesidad no está bien definida. En 2016, Hatch-Stein y colaboradores, compararon la sensibilidad y especificidad de curvas de SD-IMC con CDC- IMC, concluyendo que, para los niños y adolescentes con SD el percentil 85 en las tablas de crecimiento de CDC-IMC es un mejor indicador de exceso de adiposidad comparado con el percentil 85 en las tablas de crecimiento de SD-IMC28.
Por lo anterior, las gráficas de CDC-IMC deben ser el método para la identificación temprana de la obesidad en niños con SD 28. Esta recomendación ha sido reafirmada por la AAP estableciendo que se recomienda usar las gráficas de CDC para el IMC en niños de 10 años o más con SD 28. Adicional al seguimiento y detección oportuna de obesidad, es importante incluir una estrategia educativa para los padres en la cual se logren conocer los riesgos de esta comorbilidad e instaurar estilos de vida saludables 27.
Pubertad y fertilidad
Las mujeres con SD suelen tener una progresión puberal usual con una edad de menarca similar a la población general, con una media de 13,2 años 19. Existen varios casos de maternidad y se considera que las mujeres con SD son fértiles, por lo cual es importante la consejería anticonceptiva 29. El inicio de menopausia es más temprano con una media de presentación a los 47,1 años 11.
Por otro lado, los hombres con SD suelen presentar un desarrollo puberal usual pero con insuficiencia gonadal que conlleva niveles de testosterona normales pero hormona folículo estimulante (FSH) aumentada y bajos niveles de hormona antimülleriana18,29. Como consecuencia del hipogonadismo hipergonadotrópico es frecuente la presencia de micropene, criptorquidia, disminución del volumen testicular y de la espermatogénesis con infertilidad secundaria. No obstante, existen 3 reportes de casos de paternidad en hombres con SD y por tanto siempre se debe recomendar la anticoncepción en todo paciente postpuberal 30.
Patología tiroidea
La patología tiroidea tiene una alta frecuencia en los pacientes con trisomía 21, con una prevalencia de hasta el 66% 31. El espectro de la enfermedad es amplio e incluye hipotiroidismo (subclínico, congénito, adquirido) e hipertiroidismo (ver figura 3)13. Siendo más frecuente la presencia de hipotiroidismo2. Algunas características típicas de la patología tiroidea en SD es que no tiene predominio por sexo, no está relacionada con antecedentes familiares, con mayor frecuencia se asocia con otras enfermedades autoinmunes y suele presentarse a una edad más temprana31. La tabla 3 describe los diferentes hallazgos clínicos presentes en estos casos.
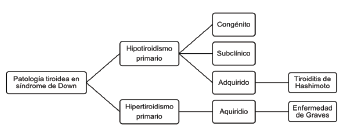
Elaboración propia a partir de esta fuente:
1Alteraciones endocrinológicas en el síndrome de Down. Rev Esp Pediatr 2012; 68(6): 440-444
Figura 3 Patología tiroidea en síndrome de Down
Tabla 3 Signos y síntomas característicos de la patología tiroidea
| Hipotiroidismo | Hipertiroidismo |
| Cansancio, somnolencia | Insomnio |
| Intolerancia al frío | Intolerancia al calor |
| Piel seca áspera o fría | Piel caliente y sudorosa |
| Estreñimiento | Diarrea |
| Lentitud motora | Hiperactividad |
| Uñas y pelo frágil | Pérdida de peso |
| Disfonía | Exoftalmos |
| Macroglosia | Dermatopatía |
| Bocio | Bocio |
Fuente: Alteraciones endocrinológicas en el síndrome de Down. Rev Esp Pediatr 2012; 68(6): 440-444.
Igualmente, es importante destacar que, aunque la expresión fenotípica se suele inclinar hacía la presencia de hipotiroidismo o hipertiroidismo, es conocido que los niños con SD pueden presentar una metamorfosis en su espectro clínico debido a las particularidades autoinmunes en este grupo de pacientes. Es decir, que podrían fluctuar entre fases de hipotiroidismo e hipertiroidismo 32.
Hipotiroidismo primario
La AAP recomienda la tamización usual de hipotiroidismo congénito junto una medición adicional de hormona estimulante de tiroides (TSH) a las 3 a 4 semanas de vida 33. Posteriormente se deberá realizar valoración de TSH a los 6 meses, a los 12 meses y a continuación de forma anual o antes en caso de presentar clínica compatible con patología tiroidea. Se recomienda que la interpretación de TSH y tiroxina libre (T4L) se realice según valores de referencia acorde a la edad 34.
Es importante la tamización de la función tiroidea en SD, pues la suplencia hormonal temprana se asocia con mejores desenlaces en el crecimiento y en el desarrollo neurológico35,36. El manejo consiste en suplencia oral con levotiroxina a dosis según la edad y el peso del paciente (ver tabla 4). La meta del tratamiento es establecer un valor de TSH normal y un valor de T4L en la mitad superior de referencia. El seguimiento se realiza como se plantea en la tabla 4 37.
Tabla 4 dosis y seguimiento de hipotiroidismo primario
TSH: hormona estimulante de tiroides. T4L: tíroxina libre.
Fuente: Congenital Hypothyroidism: A 2020-2021 Consensus Guidelines Update. Thyroid. 202131(3)387-419. Amr NH. Thyroid disorders in subjects with down syndrome: An update. Acta Biomed. 2018;89(1):132-9.
A continuación, se describen algunas características propias según la clasificación de hipotiroidismo.
Hipotiroidismo congénito
Se estima que la prevalencia de hipotiroidismo congénito (HC) en el SD es de 28 a 35 veces mayor que la de la población general y por medio del tamizaje neonatal se detecta 1 de cada 2000 a 3000 nacidos vivos 37. Datos de 2013 publicados por Cebeci y colaboradores, describen que en la mayoría de los casos el hipotiroidismo es permanente, especialmente por hipoplasia tiroidea (83% de los pacientes evaluados) 38. Sin embargo en 2020, Calcaterra y colaboradores a partir de un análisis retrospectivo de 91 niños con SD y tamización de función tiroidea, indican que el 30.8% de los niños con SD e HC tenían hipoplasia tiroidea. Además, en todos los pacientes en los que se diagnosticó HC antes de los 6 meses de edad pero con tamizaje neonatal normal, se observó hipoplasia glandular 36,39. Incluso, dado que la ecografía es un examen no invasivo, ampliamente disponible, de bajo costo y que no utiliza radiación ionizante, Cebeci y colaboradores, recomiendan la evaluación con ecografía tiroidea al nacer en todos los niños con SD 38.
La relación entre HC y SD podría estar explicada por la afección de los factores de transcripción tiroideo 1 y 2 [TTF1 - TTF2], el receptor de tirotropina [TSHR], y el factor de transcripción relacionada con locus 5 [NKX2.5]. Otro factor asociado es la posibilidad de sobreexpresión del gen DYRK1A33.
Hipotiroidismo adquirido
La principal causa es autoinmune por tiroiditis de Hashimoto y constituye una de las comorbilidades endocrinológicas más frecuentes en SD, dado que se presenta hasta en el 50% de los pacientes8,40,41. La desregulación del sistema inmune en el SD predispone a varias enfermedades de origen autoinmune y se han propuesto varias teorías como variaciones genéticas en el cromosoma 21 asociadas con la función inmune; alteraciones en la regulación de citocinas pro y antiinflamatorias; atrofia tímica con reducción de linfocitos T y B en el período neonatal, adicional a una alteración en el gen regulador autoinmune (AIRE) ubicado en 21q22.3 42. Este gen juega un papel primordial en establecer un balance entre autorreactividad e inmunorregulación y en el SD su expresión está disminuida favoreciendo el fenotipo autoinmune 42.
Las manifestaciones clínicas no difieren de los pacientes sin SD (ver tabla 4). Sin embargo, muchas veces pueden no ser detectadas o confundidas con fenotipo usual del SD, lo cual dificulta la detección de la enfermedad y tiene impacto negativo en el desarrollo cerebral y en el crecimiento 41. Adicionalmente es más frecuente en aquellos con otro tipo de patología autoinmune, como diabetes mellitus (DM), enfermedad celiaca, alopecia, vitíligo y artritis idiopática 12,28.
Hipotiroidismo subclínico
El hipotiroidismo subclínico hace referencia a la elevación de TSH con niveles de T4L normales, también denominado hipertirotropinemia aislada. Se estima una prevalencia entre 25 al 60%, pudiendo ser el tipo de patología tiroidea más frecuente en SD 37. Varios estudios plantean que la hipertirotropinemia es un atributo innato en la trisomía 21 43. El curso natural de la hipertirotropinemia no es consistente en todos los pacientes y por eso las recomendaciones de iniciar suplencia hormonal son debatidas, pues hasta un 70% de los casos puede tener resolución espontánea y la incidencia de conversión a hipotiroidismo es menor del 50% 37.
En los casos de hipotiroidismo subclínico el tratamiento solo está indicado si el valor de TSH se encuentra mayor o igual a 10 mUI/L, ante la presencia de bocio, la disminución de velocidad de crecimiento, en aquellos menores de 3 años y algunos autores suman los casos de anticuerpos antitiroideos positivos (anti-tiroglobulina y/o anti-tiroperoxidasa)43.
Hipertiroidismo primario
La prevalencia de hipertiroidismo en niños con SD oscila entre 3-6%, mucho mayor al comparar con la población general. La principal etiología del hipertiroidismo es la enfermedad de Graves 44. Dado que el hipertiroidismo presenta un cuadro sintomático agudo (ver tabla 4) comparado con los síntomas insidiosos del hipotiroidismo, el diagnóstico usualmente se realiza posterior a la sospecha clínica más que a la tamización rutinaria descrita previamente 31,44.
El tratamiento no difiere con respecto a pacientes sin SD. Las opciones terapéuticas incluyen el manejo farmacológico y la terapia definitiva (yodo ablación o tiroidectomía). El enfoque del tratamiento más utilizado es mantener el manejo farmacológico hasta por 2 años y evaluar si ha ocurrido la remisión espontánea. En caso contrario en que no haya remisión, se deberá considerar terapia definitiva 45. Es usual que en SD se requiera tratamiento definitivo 44,45.
El manejo inicial consiste en uso de medicamentos antitiroideos (metimazol, propiltiuracilo) junto con betabloqueadores, los cuales se utilizan como coadyuvantes para controlar las manifestaciones de la tirotoxicosis mientras se logra la normalización de las hormonas tiroideas 45. Dentro de opciones de antitiroideos, el metimazol es el tratamiento de primera línea a una dosis inicial de 0,2 a 0,8 mg/kg/ día. No se recomienda el uso de propiltiuracilo en niños por el alto riesgo de hepatotoxicidad 45.
Diabetes mellitus
Los niños con SD presentan una incidencia de diabetes mellitus tipo 1 (DM1) cuatro veces mayor que la población general, con un inicio más temprano de la enfermedad (22% a los 2 años contra un 7% de población general) 15. Se ha encontrado un aumento en la tasa de auto-anticuerpos relacionados con diabetes en SD a pesar de una susceptibilidad de HLA disminuida 13,46. Aitken y colaboradores, mostraron una alta prevalencia de anticuerpos antiislotes en la población con SD y una mayor coocurrencia de trastornos autoinmunitarios con diabetes y patología tiroidea en el 74%, y enfermedad celíaca y diabetes en el 14% de su cohorte 46. La clínica de la DM1 no difiere de los demás niños, los síntomas destacan polidipsia, poliuria, enuresis, pérdida de peso, polifagia (aunque en algunos casos anorexia) y finalmente debut con cetoacidosis diabética 47.
A pesar de la asociación de diabetes y SD, no está indicada la tamización sistemática de DM1. Es importante que los profesionales tengan un alto grado de sospecha clínica para de estar forma realizar un estudio temprano y evitar complicaciones agudas de la DM. Una vez concluido el diagnóstico, el tratamiento no difiere de la población general con multidosis de insulina en esquema basal-bolo47. Aunque algunos autores describen que los regímenes de insulina podrían ser más simplificados y que usualmente existe un mayor control glucémico con menos frecuencia de complicaciones vasculares (retinopatía y nefropatía) pero con mayor requerimiento de supervisión de cuidadores 2.
Con el paso de los años, también se ha descrito mayor prevalencia de resistencia a la insulina y disfunción de células B pancreáticas, que conlleva a aparición de diabetes mellitus tipo 2 (DM2) en aquellos niños con SD y sobrepeso/obesidad. Se establece la importancia de instaurar pautas de estilo de vida saludable y en los casos en que existan signos clínicos de resistencia a insulina, como acantosis nigricans, sospechar de una posible DM2 48.
Salud ósea
Diversos factores tienen un impacto negativo en la salud ósea de pacientes con SD, dentro de los cuales se destacan la hipotonía, la vida sedentaria, la obesidad, las comorbilidades frecuentes (como malabsorción), la presencia de convulsiones con uso de anticonvulsivante y las dificultades nutricionales asociadas a una deficiencia e insuficiencia de vitamina D. Se ha demostrado que adultos y jóvenes con SD tienen una densidad mineral ósea disminuida, aunque aún se requieren estudios que evidencien el riesgo real de fracturas 17,49-51. Por esta razón, para la prevención primaria de las fracturas osteoporóticas en adultos con SD, no hay pruebas suficientes para recomendar a favor o en contra de la aplicación de pautas establecidas para detección de osteoporosis, incluida la estimación del riesgo de fractura o la realización de densitometría ósea. En cambio, todos los pacientes con SD que sufran una fractura por fragilidad, deben ser evaluados por causas secundarias de osteoporosis, incluida la detección de hipertiroidismo, enfermedad celíaca, deficiencia de vitamina D, hiperparatiroidismo y medicamentos asociados con efectos adversos en la salud ósea 52.
Actualmente, la recomendación durante la infancia es reforzar el adecuado consumo de calcio y vitamina D, exposición solar y establecer un estilo de vida más activo que permita mejorar el tono y fuerza muscular. Estas intervenciones demuestran el aumento de masa ósea en jóvenes con SD. Solo se sugiere realizar evaluación de laboratorios con calcio, fósforo y 25hidroxi-vitamina D en aquellos con factores de riesgo adicional como el uso de anticonvulsivante, antecedente de patología oncológica, entre otros53,54.
Alteraciones en el metabolismo de lípidos
Previamente se consideraba al SD como un modelo de "enfermedad libre de aterosclerosis", sin embargo, estudios más recientes sugieren un aumento del riesgo de mortalidad por enfermedad cardiovascular isquémica y cerebrovascular en pacientes con SD comparado con la población general 55. El mecanismo fisiopatológico estaría relacionado con alteraciones en el perfil lipídico, sumado a la alta incidencia de cardiopatía congénita, patología tiroidea y sedentarismo que favorecen el aumento del riesgo cardiovascular 55,56.
Pocos son los estudios que evalúan el perfil lipídico de pacientes con SD y sus resultados son conflictivos55. Por ejemplo, un estudio realizado por Adelekan y colaboradores en un centro de referencia de enfermedades metabólicas en Estados Unidos obtuvo el perfil lipídico de 27 niños con SD entre 4-10 años y como control contó con 31 hermanos sanos. Al comparar los resultados de los dos grupos ninguno requirió intervención nutricional o médica adicional. Aunque las diferencias no fueron estadísticamente significativas, la media de los niveles de colesterol total, colesterol de baja densidad (LDL) y triglicéridos fue mayor en niños con SD que el grupo control, junto con niveles de colesterol de alta densidad (HDL) más bajos 56.
Aún no existe una recomendación en guías sobre el monitoreo de perfil lipídico en SD y tampoco existe evidencia suficiente que permita establecer un objetivo terapéutico o indicación de manejo hipolipemiante diferencial 57. Se requieren estudios longitudinales que relacionen los hallazgos de perfil lipídico con morbilidad y mortalidad cardiovascular para definir si se trata de un grupo de alto riesgo y de esta forma establecer objetivo terapéutico diferencial 58. Por ahora, se insiste en la importancia del monitoreo de IMC y de implementar estilos de vida saludable 57,58.
En la tabla 5 se realiza un resumen de las complicaciones endocrinológicas asociadas al SD, con sus diferentes consideraciones en cuanto a seguimiento y tratamiento.
Tabla 5 Recomendaciones en el seguimiento y tratamiento de endocrinopatías asociadas al SD
TSH: Hormona estimulante de tiroides. T4L: Tiroxina libre. LDL: Colesterol de baja densidad. HDL: Colesterol del alta densidad
Elaboración propia a partir de estas fuentes:
1 Health Supervision for Children With Down Syndrome. Pediatrics August 2011,128 (2) 393-406
2 Down's Syndrome Association (DSA): Annual health check guideline (2015)
3 Clinical practice guidelines for management of children with Down syndrome. Journal of Pediatric Health Care (2014)
Conclusión
El síndrome de Down constituye la cromosopatía más frecuente y dentro de sus complicaciones resaltan las patologías endocrinológicas. Por lo cual, es importante el abordaje interdisciplinario del paciente pediátrico con SD, que incluya al endocrinólogo pediatra. Estas incluyen alteraciones en crecimiento, que están presentes en casi todos los pacientes y presentan un patrón de crecimiento característico que implica el uso de curvas adecuadas para evaluar la condición. El desarrollo de obesidad es otro parámetro para considerar y que debe ser prevenido, dado que su frecuencia puede ser tan alta como del 20%. Otras complicaciones son resultado de la alta prevalencia de patología autoinmune, como es la patología tiroidea, en especial el hipotiroidismo, evidenciado hasta en 66% de los casos y que requiere un seguimiento estricto del perfil tiroideo. Es importante educar a los padres sobre signos típicos de DM y siempre recordar que existe la posibilidad de fertilidad en este grupo de pacientes, por lo cual la consejería anticonceptiva siempre debe hacer parte de la valoración integral. El reconocimiento adecuado de las complicaciones endocrinológicas es vital, dado que permite establecer un abordaje integral y preventivo en el paciente con SD. Sin embargo, aún existen vacíos en la literatura sobre aspectos como la salud ósea y las dislipidemias, por lo que se requiere un mayor número de estudios que permitan realizar recomendaciones médicas basadas en la evidencia.

