










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.15 no.1 Medellín Jan./Mar. 2002
INVESTIGACIÓN ORIGINAL
Hepatopatía crónica en niños. Justificación para un programa de transplante hepático infantil
MARÍA ELSY SEPÚLVEDA HINCAPIÉ*; NORA LUZ YEPES PALACIO*; FERNANDO ALBERTO GUTIÉRREZ MENDOZA**
* Docente de Pediatría. Departamento de Pediatría y Puericultura. Universidad de Antioquia. Medellín, Colombia.
** Médico Patólogo. Clínica Las Vegas. Medellín, Colombia.
LOS NIÑOS PUEDEN SER AFECTADOS por varias enfermedades que a mediano o largo plazo producen falla hepática tanto aguda como crónica. El transplante del hígado es la única opción terapéutica disponible para estos casos. En todos los centros de transplante de hígado, la principal indicación para esta intervención en los niños es la atresia de vías biliares; esta enfermedad se manifiesta desde el período neonatal y el transplante puede ser necesario tan temprano, como entre los nueve y dieciocho meses de edad. Otras enfermedades de aparición más tardía pueden necesitar del transplante en etapas posteriores de la vida.
Se presenta la casuística de niños con enfermedad hepática crónica tributaria de transplante hepático, en los niños atendidos en un servicio especializado de tercer nivel en Medellín, Colombia, durante el periodo comprendido entre enero de 1990 y diciembre de 1996.
PALABRAS CLAVE
HÍGADO, TRANSPLANTE, NIÑOS, HEPATOPATÍA CRÓNICA, TRANSPLANTE HEPÁTICO
SUMMARY
CHILDREN MAY BE AFFECTED BY VARIOUS DISEASES that can result, at short or long term, in acute or chronic liver failure. Liver transplant is the only therapeutic option available in these patients. In most major transplant centers, biliary atresia is the main indication for liver transplantation in children. Children affected with biliary atresia become symptomatic in the neonatal period and may require transplantation as early as at 18 months, while other diseases that manifest later in life require transplantation at later ages.
In this report we present our experience with chronic liver diseases in childhood that required transplantation in a specialized service located in a third level hospital in Medellín, Colombia, over a period of 6 years (January 1990 to December 1996).
INTRODUCCIÓN
EL PRIMER TRANSPLANTE DE HÍGADO EN HUMANOS lo hizo Thomas Starzl (1) en 1963, a una niña con atresia de vías biliares, posteriormente, en 1983, el gobierno de Estados Unidos lo aprobó como terapia. Desde entonces se han logrado avances importantes, no sólo en el perfeccionamiento de las técnicas quirúrgicas, que han permitido la utilización de diferentes tipos de transplantes como el total de cadáver, los segmentarios de cadáver y los reducidos de donantes vivos relacionados (2,3), sino también en el tratamiento médico, en el que resalta el uso de inmunosupresores más seguros (4), lo que logra mejorar el tiempo y la calidad de vida de los pacientes que han recibido un transplante de hígado.
Este es una terapia aprobada para el tratamiento definitivo de niños y adultos que sufren enfermedades hepáticas severas, agudas o crónicas, que producen insuficiencia o falla hepática. Un alto porcentaje de estos transplantes se hacen en niños y en ellos, el 50% tienen diagnóstico de atresia de vías biliares (5), enfermedad que se manifiesta con una colestasis neonatal. Su tratamiento definitivo es el transplante del hígado y se debe hacer entre los 9 y 18 meses de edad cuando el tratamiento inicial, la portoenteroanastomosis o cirugía de Kasai, no pudo hacerse o no se lograron los resultados esperados, como es obtener un drenaje adecuado de la bilis.
Existen además otras causas de colestasis neonatal, como la deficiencia de alfa1 antitripsina, la hepatitis neonatal producida por el sindrome de TORCHS, (toxoplasmosis, rubéola, enfermedad citomegálica, herpes y sífilis) u otras enfermedades hepáticas de la infancia que pueden necesitar el transplante en un período de evolución más tardío (6).
Con este informe se presenta la casuística correspondiente a los niños atendidos en un servicio de Gastroenterología Infantil, en la ciudad de Medellín, durante el periodo de seis años, comprendido entre enero de 1990 y Diciembre de 1996, teniendo en cuenta que ellos necesitaran un transplante hepático ya fuera a mediano o a largo plazo.
MATERIALES Y MÉTODOS
ESTE ES UN ESTUDIO DESCRIPTIVO Y RETROSPECTIVO, en el que se estudiaron las características clínicas de los pacientes, la edad al inicio de los síntomas y en el momento de la primera evaluación; el diagnóstico comprobado, su pronóstico y evolución en relación con la indicación para el transplante hepático. Los datos se tomaron de la historia clínica y el diagnóstico se estableció con base en los hallazgos clínicos, los resultados de los exámenes de laboratorio y del estudio histológico.
RESULTADOS
EL GRUPO ESTUVO INTEGRADO POR 79 PACIENTES, de los cuales 49 fueron niños (62%) y 30 fueron niñas (48%). Se dividieron en dos grupos; el primero, estuvo integrado por 68 pacientes cuyo diagnóstico fue de colestasis neonatal, y el otro, constituido por 11 pacientes que sufrieron hepatopatías diferentes de la colestasis neonatal (Tabla 1).
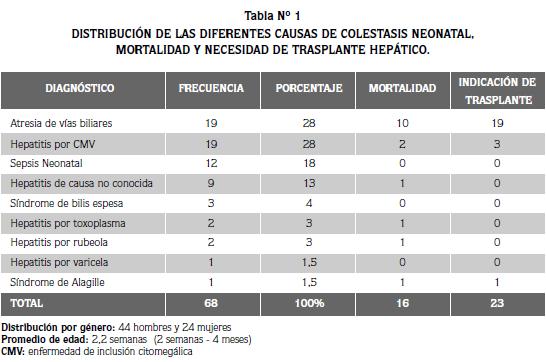
En el grupo de 68 pacientes cuyo diagnóstico fue colestasis neonatal, la distribución según las causas fue la siguiente: 19 niños (28%) con atresia de vías biliares (AVB), 19 (28%) con hepatitis neonatal causada probablemente por enfermedad de inclusión citomegálica (CMV), y 30 (44%), que sufrieron colestasis de diferentes etiologías. Del grupo total, el 34% (23/68) fueron candidatos a transplante hepático: los 19 niños con atresia de vías biliares, los tres que padecieron hepatitis neonatal producida por la enfermedad de inclusión citomegálica, y uno con síndrome de Alagille.
Al momento del seguimiento correspondiente a los controles para esta revisión, sólo dos niños con AVB habían recibido su transplante en instituciones diferentes: una niña que falleció ocho semanas después del transplante y un niño que había sobrevivido cinco años y se encontraba en buenas condiciones. Diez niños que tuvieron atresia de vías biliares (AVB), fallecieron a causa de la enfermedad antes de recibir el transplante, mientras que los siete restantes, han continuado enfermos y esperan un transplante que se hará en un tiempo variable según su evolución.
EL promedio de edad al momento de la remisión y evaluación por especialista fue de 10 semanas ( rango de 2 a 16 semanas), es una edad tardía si se tiene en cuenta que en la AVB los resultados son más satisfactorios cuando la derivación se hace entre la sexta y octava semanas de vida. Todos los niños tuvieron como manifestaciones clínicas la ictericia con acolia y coluria, estos hallazgos no hacen diferencia entre las diferentes causas de colestasis neonatal y se consideran altamente sugestivos de atresia de vías biliares, solamente los exámenes bioquímicos, de imaginología y la biopsia aún con laparotomía exploradora permiten establecer el diagnóstico definitivo
Los otros 30 niños con colestasis neonatal, no se consideraron candidatos al transplante; tres de ellos fallecieron debido a la gravedad de las alteraciones neurológicas concomitantes ya que sus diagnósticos fueron: síndrome de rubéola congénita, toxoplasmosis congénita y hepatitis neonatal de causa no aclarada. Los otros veintisiete se recuperaron de su hepatopatía y no requirieron del trasplante.
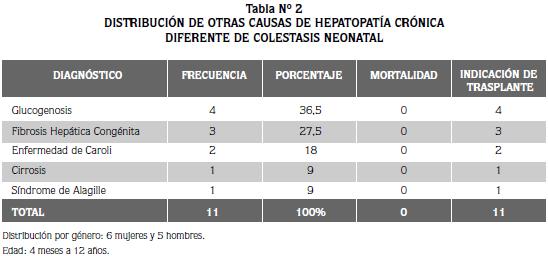
En el segundo grupo, compuesto por 11 pacientes, la enfermedad hepática se manifestó después de la etapa neonatal; todos son candidatos al transplante hepático en el futuro y sus diagnósticos son: tres casos de fibrosis hepática congénita, cuatro de glucogenosis, dos de enfermedad de Caroli y sendos casos de cirrosis y síndrome de Alagille. En este grupo de niños, la edad al momento de la evaluación fue muy variable, promedio de 4 años y rango entre 4 meses y 12 años.
El total de niños con hepatopatía crónica candidatos a transplante hepático es de 34/79 que corresponde al 43 % (Tabla 2). De ellos el 55% (19/34), corresponde al diagnóstico de atresia de vías biliares.
DISCUSIÓN
AUNQUE LA ATRESIA DE VÍAS BILIARES es la indicación más frecuente de trasplante de hígado en niños, existen otras enfermedades hepáticas diferentes en su origen y evolución que requieren este tratamiento. El momento oportuno para el trasplante está determinado por la magnitud de la falla hepática producida por la evolución progresiva de la enfermedad, esta falla se define teniendo en cuenta pruebas de laboratorio que evalúan la capacidad de síntesis hepática o reserva funcional: albúmina sérica, concentración sérica de bilirrubina, tiempo de protrombina, tiempo parcial de tromboplastina, colesterol sérico y algunas manifestaciones clínicas como ascitis, antecedente de encefalopatía hepática y el estado nutricional (7, 8).
La colestasis neonatal es un síndrome de frecuente en nuestro medio, sus causas son variadas y de gravedad diferente. Algunas son enfermedades con pronóstico favorable, pero otras son progresivas y requieren terapia agresiva y oportuna como el trasplante hepático. El 50% de los trasplantes en pediatría, se hacen en niños con diagnóstico de atresia de vías biliares; la necesidad del trasplante puede ser temprana, antes de los dos años de edad, en aquellos pacientes en quienes la cirugía de Kasai, derivación biliodigestiva o portoenteroanastomosis, no logra el flujo de bilis adecuado o cuando no es posible hacerla. En otras ocasiones cuando la derivación biliodigestiva funciona, produciendo drenaje adecuado de bilis al intestino por un tiempo variable, la necesidad de trasplante, el tratamiento considerado definitivo, es más tardía pero finalmente se llega a él (9,10).
El momento de hacer el trasplante está determinado por la evolución de cada paciente, esta es variable, y depende de varios factores como la edad a la que se hizo la derivación, mientras más precoz, entre la sexta y octava semana de vida, mayor es la posibilidad de éxito, del tipo de atresia, de la respuesta a la derivación biliodigestiva, del antecedente de colangitis, la aparición de complicaciones como hipertensión porta, y además de la disponibilidad de donadores de injertos. En esta casuística la atresia de vías biliares fue una de las principales causas de colestasis neonatal, 19/68, a pesar de la derivación biliodigestiva, los pacientes evolucionaron con alteración progresiva de la función hepática, diez fallecieron, siete son candidatos a trasplante y dos fueron trasplantados en otras instituciones pero uno falleció dos meses después del trasplante.
Otras causas de colestasis neonatal son las infecciones por enfermedad de inclusión citomegálica, toxoplasmosis, rubeola, sífilis, virus del herpes, constituyendo el síndrome de TORCH, además de la sepsis neonatal y otras enfermedades metabólicas, infecciosas y aún de causa no aclarada (11).
Para establecer el diagnóstico de hepatitis neonatal por enfermedad de inclusión citomegálica, se requiere además de la sintomatología clínica, determinar la presencia del virus por cultivo o antigenemia (12); al momento de la evaluación de los pacientes no se disponía de estas pruebas, y para sustentar el diagnóstico en los casos descritos, se tuvo en cuenta el resultado de la determinación de los anticuerpos específicos tipo IgM e IgG tanto en la madre como en el bebé, comparando las cifras de los anticuerpos tipo IgM y tomando como dato positivo, que la concentración de IgM fuera más alta en el niño que en la madre. Sin embargo, es necesario tener en cuenta que no toda infección indica enfermedad y para considerar la infección como perinatal, esta debe ser detectada antes de las cuatro semanas de vida. En los pacientes clasificados con diagnóstico probable de enfermedad de inclusión citomegálica, se descartaron otras causas de enfermedad hepática colestásica temprana, sin encontrar indicios de otra enfermedad diferente que explicara su hepatopatía y la determinación de anticuerpos fue compatible. Los hallazgos histológicos descritos en colestasis neonatal como tapones biliares, necrosis hepatocelular, colestasis intracelular, eritropoyesis extramedular, células gigantes, proliferación o disminución de conductos biliares, fibrosis intercelular y cirrosis, son inespecíficos y sólo sugieren la causa de la colestasis. Cuando hay predominio de algunos elementos como la proliferación de conductos con tapones biliares, la colestasis intracelular y la fibrosis estos apoyan el diagnóstico de atresia de vías biliares. La necrosis celular con infiltrado inflamatorio, la eritropoyesis extramedular y la degeneración celular sugieren otras causas (13).
Las células gigantes con cuerpos de inclusión, son poco frecuentes y su presencia se considera característica de la enfermedad por CMV, pero no fueron detectadas en ninguno de los casos. La frecuencia de otras causas de colestasis neonatal en esta casuística, se refieren en la tabla N° 2.
El síndrome de Alagille, al igual que otras colestasis como cirrosis biliar primaria, colangitis esclerosante, enfermedad de Caroli, fibrosis hepática congénita variedad colangítica, producen colestasis crónica, pueden evolucionar a cirrosis y algunas aún a hepatocarcinoma (14). Cuando se presentan las consecuencias clínicas de la colestasis tales como fracturas, prurito no controlable, hipercolesterolemia, y coagulopatía que no mejoran a pesar de tratamiento médico, se indica el trasplante hepático (15); requieren pues vigilancia y tratamiento permanentes para establecer oportunamente la necesidad de esta terapia definitiva. Este síndrome se diagnosticó en dos niños, uno en la época de lactante y evolucionó con afectación progresiva del hígado que le causó cirrosis, y murió sin tener oportunidad de recibir el trasplante. El otro niño, se diagnosticó en edad escolar y al momento de esta evaluación, continuaba sintomático pero sin signos de falla hepática.
Las otras hepatopatías fueron menos frecuentes, pero no por ello menos importantes, la morbilidad y mortalidad que presentan a largo plazo, son retos que pueden ser superados ofreciendo asistencia adecuada y acorde con el desarrollo científico actual, sin embargo en etapa tardía pueden ser indicación de trasplante.
En el grupo de las glucogenosis, el trasplante está indicado en el tipo Ia y Ib cuando se complican con adenoma y en las glucogenosis tipo III, IV y VI (16), cuando a pesar de tratamiento médico evolucionan a cirrosis, hipertensión porta de difícil control, u otra complicación no controlable.
Al igual que en otros países, el diagnóstico de atresia de vías biliares correspondió al mayor porcentaje 55% de pacientes candidatos a trasplante hepático 19/34. Llama la atención que en esta casuística no se tuvo ningún paciente con diagnóstico de enfermedad metabólica tipo galactosemia, deficiencia de alfa uno antitripsina y tirosinemia, enfermedades que son reportadas en otros países como causa de colestasis o hepatopatía temprana.
En países desarrollados el trasplante hepático es un tratamiento que se utiliza desde hace varios años tanto en niños como en adultos, con resultados favorables y alentadores que han permitido avanzar en su investigación mejorando las probabilidades de sobrevivir con calidad de vida adecuada (17). En nuestro medio existen enfermedades cuya frecuencia y gravedad justifican la organización de entidades y grupos de trabajo dedicados a ofrecer esta terapia, es pues una necesidad que debe tenerse en cuenta y amerita ser atendida.
BIBLIOGRAFÍA
1. STARZL TE, MACHIORO TL, VON KAULLA KN, HERMANN G, BRITTAIN RS, WADDELL WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117: 659-676. [ Links ]
2. EMOND JC, WHITINGTON PF, THISTLETHWAITE JR, ALONSO EM, BROELSCH CE. Redicud-size orthotopic liver transplantation: Use in the management of children with chronic liver disease. Hepatology 1989; 10: 867-872. [ Links ]
3. HEFFRON TG. Living-related pediatric liver transplantation. Sem Pediatr Surg 1993; 2: 248-53. [ Links ]
4. WHITINGTON PF, BALISTRERI WF. Liver transplantation in pediatrics: indications, contraindications and pretransplant management. J Pediatr 1991; 118: 169-177. [ Links ]
5. MURRAY KF, JONAS MM. Biliary atresia. En: Altschuler SM, Liacouras CA editores. Clinical Pediatric Gastroenterology. Philadelphia: Churchill Livingstone; 1998: 331-339. [ Links ]
6. ROSENTHAL P, PODESTA L, SHER L, MAKOWAKA L. Liver transplantation in children. Am J Gastroenterol 1994; 28: 480-492. [ Links ]
7. MALATACK JJ, SCHAID DJ, URBACH AH, GÄRTNER JC, ZITELLI BJ, ROCKETTE H, et al. Choosing a pediatric recipient for orthotopic liver transplantation. J Pediatr 1987; 111: 479-489. [ Links ]
8. PUGH RW, MURRAY-LYON IM, DAWSON JL. Transection of the esophagus for bleeding esophageal varices: final analysis of a controlled trial. Br J Surg 1983; 60: 646-449. [ Links ]
9. BALISTRERI WF, GRAND R, HOOFNNAGLE JH, SUCHY FJ, RYCKMAN C, PERLMUTTER DH, et al. Biliary atresia: Current concepts and research directions. Summary of a symposium. Hepatology 1996; 23: 682-692. [ Links ]
10. DAVENPORT M, KERKAR N, MIELI-VERGANI G, MOWAT AP, HOWARD ER. Biliary atresia: The King's College Hospital Exprience (1974-1995). J Pediatr Surg 1997; 32: 479-85. [ Links ]
11. MOWAT AP, DAVIDSON LL, DICK MC. Earlier identification of biliary atresia and hepatobiliary disease: selective screening in the third week of live. Arch Dis Child 1995; 72: 90-92. [ Links ]
12. TARR PI, HAAS JE, CHRISTIE DL Biliary atresia, cytomegalovirus, and age at referral. Pediatrics 1996; 97: 828-831. [ Links ]
13. LAI MW, CHANG MH, HSU SC, HSU HC, SU CT, KAO CL, et al. Differential diagnosis of extrahepatic biliary atresia from neonatal hepatitis: A prospective study. J Pediatr Gastroenterol Nutr 1994; 18: 121-127. [ Links ]
14. RABINOVITZ M, IMPERIAL JC, SCHADE RR, VAN THIEL DH. Hepatocellular carcinoma in Alagille's Sindrome: a family study. J Pediatr Gastroenterol Nutr 1989; 8: 26-30. [ Links ]
15. SOKOL RJ. Medical management of the infant or child with chronic liver disease. Semin Liver Dis 1987; 7: 155-167. [ Links ]
16. MCDIARMID SV. The liver and metabolic disease of childhood. Liver Transpl Surg 1998; 4: S34-S50. [ Links ]
17. GOSS JA, SHACKLETON CR, MCDIARMID SV, MAGGARD M, SWENSON K, SEU P, et al. Longterm results of pediatric liver transplantation. An analysis of 569 transplants. Ann Surg 1998; 228: 411-420. [ Links ]
