










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.15 no.2 Medellín Apr./June 2002
PRESENTACIÓN DE CASO
Parálisis periódica hipocalémica familiar (PPHF): reporte de un caso y revisión del tema
PERIODIC FAMILIAL HYPOKALEMIC PARALYSIS. REPORT OF A CASE AND REVIEW OF THE LITERATURE
MARGARITA MARÍA VÉLEZ C.1; JAIME CARRIZOSA M.2; WILLIAM CORNEJO O.3
1 DOCTORA, Residente III Nivel de Pediatría, Universidad de Caldas
2 DOCTOR, Neurólogo Infantil, Universidad de Antioquia, Medellín, Colombia
3 DOCTOR. Neurólogo Infantil, Universidad de Antioquia, Medellín, Colombia.
LA PARÁLISIS PERIÓDICA HIPOCALÉMICA FAMILIAR es una enfermedad que pertenece al grupo de las canalopatías. Consiste en la presentación de episodios de parálisis muscular progresivos en intensidad y frecuencia acompañada de hipocalemia. Dos mutaciones explican la presencia de la enfermedad, la CACNA1S y la SCN4A, que afectan los canales de potasio mediados por calcio y los canales de sodio, respectivamente. Lo anterior repercute en la función de los canales para el potasio mediados por voltaje, llevando a una hipocalemia extracelular sostenida produciendo despolarización continua con parálisis. Los desencadenantes son el ejercicio, los carbohidratos, el frío, el estrés, entre otros, y las pruebas de provocación clínica se realizan con insulina, glucosa y ejercicio. La enfermedad se puede prevenir evitando estos factores y tiene alguna respuesta al tratamiento con acetazolamida. Se presenta el caso clínico de un niño que cumple con los criterios clínicos de paresia asociada a hipocalemia desencadenada por carbohidratos.
PALABRAS CLAVE
HIPOTONÍA, PARÁLISIS, HIPOCALEMIA, MUTACIÓN
SUMMARY
Periodic hypokalemic familial paralysis (PHFP) is a channel-mediated disease. Increasing focal or generalized muscular paralytic episodes are associated with low serum potassium levels. Two point mutations are described, CACNA1S and SCN4A. These mutations affect calcium and sodium mediated potassium channels. A continuous depolarization with low extracelular potassium promotes hypotonia. Known stressors are excercise, cold, stress and high carbohydrate intake. Control of stressors and azetazolamyde are the main treatment options. We describe one patient with clinical and laboratory characteristics of PHFP.
REPORTE DEL CASO
ESTE ES UN PACIENTE DE SEXO MASCULINO de 2 años y 10 meses de edad, sano desde el nacimiento, quien a los 2 años y 7 meses presentó un primer episodio de dolor abdominal, astenia, adinamia, dolor en las extremidades inferiores, hipotonía y debilidad generalizada que le imposibilitaba la marcha y la sedestación. Todo lo anterior ocurrió al día siguiente de una alta ingesta de carbohidratos con pastel y helado por una fiesta de cumpleaños. El paciente consultó al servicio de urgencias y fue tratado con líquidos endovenosos con recuperación total del cuadro clínico y readquisición de la marcha normal en las siguientes 4 horas. A los 2 meses del primer episodio nuevamente desarrolló sintomatología y signología similares, precedidas también de una alta ingesta de carbohidratos. Al examen físico de igual manera había hipotonía generalizada, imposibilidad para la marcha y arreflexia. El potasio sérico se encontró disminuido con un valor de 1,9 mmol/L. Los demás exámenes de laboratorio fueron normales: sodio, calcio y cloro séricos; gases arteriales, magnesemia y fosfemia, T3, T4 y TSH, hemoleucograma, pruebas de función hepática y renal, PCR y creatinfosfocinasa. Se trató con líquidos endovenosos y se le hizo reposición endovenosa lenta de potasio. Presentó recuperación total del cuadro a las 24 horas de evolución. Se le hicieron recomendaciones e indicaciones dietéticas sin aparición de nuevas recaídas.
Los antecedentes prenatales, perinatales, patológicos y familiares no aportaron datos relevantes. Su desarrollo psicomotor ha sido normal.
REVISIÓN
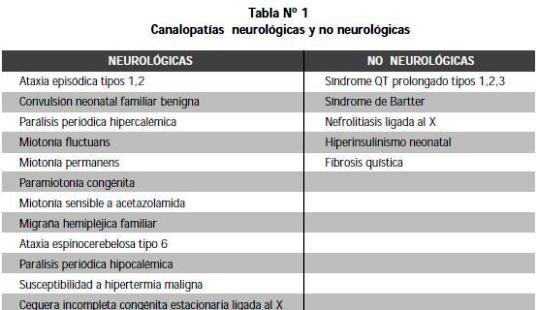
LA COMUNICACIÓN INTERCELULAR depende de señales eléctricas producidas por el paso de iones cargados, el cual es mediado por proteínas o canales que se encuentran en la membrana celular. Las enfermedades que comprometen el paso de estos iones son llamadas canalopatías (Tabla 1) (1,2).
Los canales iónicos están conformados por subunidades que configuran un poro central. Cada canal es selectivo para las sustancias que deja pasar cumpliendo el papel de compuerta. Dependiendo de la fuerza de conducción mediada por voltaje pueden pasar los diferentes iones de un lado a otro de la membrana. El potencial de acción está involucrado de una manera importante en estas enfermedades, ya que depende completamente de los canales iónicos mediados por voltaje. Las canalopatías llevan a alteración del potencial de acción y al mal funcionamiento de diferentes sistemas. El potencial de acción depende de los canales iónicos por el siguiente mecanismo: cuando hay cambios en el voltaje transmembrana se abre un canal de sodio y estos iones pasan rápidamente de una alta concentración externa al interior de la célula. La célula se despolariza, haciendo que los canales de potasio mediados por voltaje se abran y estos iones salen de la célula restableciendo el potencial de membrana en reposo. La salida de iones potasio genera un potencial negativo a través de la membrana. El flujo neto de potasio termina cuando el gradiente de concentración que hace salir iones potasio es balanceado por un gradiente eléctrico que retarda la salida de iones cargados positivamente (2-4).
Entre estas enfermedades se encuentran la parálisis periódica hipocalémica, la parálisis periódica sensible al potasio y la paramiotonía congénita. La parálisis periódica hipocalémica familiar (PPHF) es la forma más común de parálisis periódica; se encuentra una prevalencia aproximada de 1:100.000, es autosómica dominante con menor penetrancia en mujeres.
Estudios en sangre arteriovenosa han sugerido que la hipocalemia es generada por la captación de potasio dependiente de insulina desde el espacio extracelular a la fibra muscular, pero se desconoce si el número de receptores o la afinidad de la insulina están incrementados (5). También se ha encontrado que la disminución del potasio extracelular lleva a despolarización de la membrana del músculo con parálisis periódica, pero a hiperpolarización del músculo normal. El aparato contráctil de los individuos afectados es normal, al comprobarse la contracción de la fibra muscular no excitable eléctricamente con la exposición al calcio (6).
Esta enfermedad se da por una mutación en los canales de calcio del músculo esquelético, en el gen CACNL1A3 del cromosoma 1q31. Dicho gen codifica la subunidad a1 que hace parte del complejo receptor dihidropiridina /canal de calcio y es formadora de poros de los canales de calcio tipo-L mediados por voltaje. Normalmente esos poros, en una interacción con el receptor ryanodino, funcionan como un sensor de excitación-contracción activado por voltaje a través de la liberación de calcio desde el retículo sarcoplásmico (5).
Se ha sugerido que un defecto en la función de los canales de potasio dependientes del ATP del músculo esquelético contribuye al defecto, explicando por qué el ejercicio o la ingestión de carbohidratos desencadenan la parálisis. Tricarico y colaboradores (7) han demostrado la disminución de la salida de potasio a través de los canales de potasio dependientes de ATP y también la disminución de esta corriente con la administración de insulina o la hipocalemia. Se postula que hay un acoplamiento entre la Na/K ATPasa y los canales de potasio dependientes de ATP en el músculo esquelético de la siguiente manera: en las personas normales la insulina activa la Na/K ATPasa y lleva a la entrada de iones potasio y a la consecuente hipocalemia transitoria. La activación de los canales de potasio dependientes de voltaje lleva a la salida del mismo ion y a la compensación de la hipocalemia, manteniendo la hiperpolarización de la fibra muscular. En los pacientes con PPHF no ocurre la salida de potasio y por lo tanto una hipocalemia y una despolarización sostenidas de la membrana explican la presentación clínica de la enfermedad.
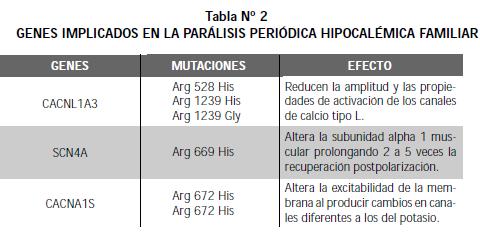
Ruff y col. (8) refieren que las fibras musculares de los individuos con la enfermedad tienen un aumento en el umbral del potencial de acción y que la fracción de las fibras excitables disminuye con el incremento de fibras despolarizadas. En su estudio encontraron que la insulina potencia la despolarización de las fibras musculares de las personas con PPHF por reducir la rectificación interna en la conductancia del potasio. Los genes implicados en la PPHF se describen en la Tabla 2 (9-14).
PRESENTACIÓN CLÍNICA
LA SINTOMATOLOGÍA comienza por lo general durante la pubertad, con rangos entre la infancia y la tercera década de la vida. En el 60% de los pacientes aparece antes de los 16 años. Inicialmente los ataques son infrecuentes y después de unos meses o años se incrementan y pueden recurrir diariamente. Los pacientes presentan paresia o parálisis muscular parcial o generalizada más comúnmente después de un ejercicio fuerte o de ingerir alimentos ricos en carbohidratos. El músculo esquelético es el más involucrado, pero con poca afección de los músculos respiratorios. No se presentan cambios en la sensibilidad ni en el estado de conciencia, y los reflejos osteotendinosos están ausentes. Durante el episodio pueden ocurrir oliguria, constipación y diaforesis. Se recobra la fuerza en espontáneamente en horas pero en ocasiones puede tardar días. Los factores desencadenantes son múltiples y se describen en la Tabla 3.

En los exámenes de laboratorio se encuentra disminución de los niveles séricos de potasio y fósforo, aunque éstos pueden estar normales. En el electrocardiograma pueden aparecer signos de hipocalemia como bradicardia y cambios en la onda T y el segmento ST y aparición de onda U.
La severidad y la evolución de la enfermedad dependen de la penetrancia genética. Algunos pacientes pueden tener resolución de los síntomas en la quinta o sexta décadas. Los pacientes que tienen la enfermedad grave pueden tener en el día varias crisis de intensidad variable, más fuertes en la noche y al amanecer. Algunos individuos pueden presentar una miopatía permanente, con debilidad residual.
Sternberg y col (14) hicieron un estudio en 58 pacientes de familias diferentes buscando la frecuencia de cada una de las mutaciones descritas y en 45 casos encontraron una mutación causante de la enfermedad, de los cuales 40 la tenían en el gen CACNA1S y cinco en el SCN4A. En los pacientes con esta última mutación encontraron una completa penetrancia en ambos sexos, edad temprana de comienzo, mialgias postcrisis y un alto número de episodios inducidos por acetazolamida. La biopsia en dos de estos pacientes mostró una miopatía caracterizada por agregados tubulares. En cambio, en los pacientes con la primera mutación, la biopsia mostró predominio de vacuolas.
EVALUACIÓN DIAGNÓSTICA
EN LOS PACIENTES CON ESTA ENFERMEDAD los niveles de potasio están usualmente disminuidos, pero cuando la disminución es exagerada o están bajos después de la crisis se debe pensar en una parálisis periódica secundaria. Los niveles de creatinfosfocinasa (CPK) están normales o un poco aumentados entre las crisis o pocos días después. Los estudios de conducción nerviosa y la electromiografía (EMG) pueden ser normales cuando el paciente es asintomático. Durante las crisis los estudios de conducción nerviosa pueden mostrar ausencia o disminución de la amplitud del potencial de acción del músculo. En la EMG se puede encontrar ausencia o disminución de la actividad de inserción con poco potencial de acción de las unidades motoras (5).
Se pueden hacer posteriormente pruebas de provocación aplicando glucosa e insulina, con monitoreo de componentes del potencial de acción del músculo. Estas pruebas no deben hacerse en pacientes con enfermedades de base como insuficiencia renal, insuficiencia adrenal, hipocalemia o tirotoxicosis. Se administran 2 g/kg de glucosa temprano en la mañana combinados con 10 a 20 unidades de insulina cristalina por vía subcutánea, lo cual puede desencadenar el ataque paralítico en 2 a 3 horas. La prueba es positiva cuando se presenta la debilidad y el potasio está por debajo de 3 mmol/L; sin embargo, existen resultados falsos negativos.
PREVENCIÓN Y TRATAMIENTO
CUANDO EL ATAQUE ES LEVE no necesita tratamiento. Cuando produce parálisis, se deben administrar 2 a 10 g de cloruro de potasio en solución acuosa por vía oral. En la mayoría de los casos se recupera la fuerza muscular en media a una hora. Si en 2 a 3 horas el paciente no ha mejorado se puede repetir la dosis. No se recomienda el potasio intravenoso porque puede llevar a una hipercalemia que puede poner en peligro la vida.
Se deben poner en práctica medidas preventivas evitando los factores desencadenantes, asociados a dietas bajas en carbohidratos y sodio y ricas en potasio.
La droga profiláctica de elección es la acetazolamida a una dosis lo más baja posible, por ejemplo 125 mg/día, pero si las crisis continúan se debe aumentar hasta una dosis máxima de 250 mg/d. En algunos individuos se ha visto que la droga puede precipitar la crisis. Los pacientes refractarios a la acetazolamida pueden responder a la diclorfenamida, otro inhibidor de la anhidrasa carbónica. Se administran 25 mg tres veces al día (15,16).
DISCUSIÓN
LA PPHF ES UNA ENFERMEDAD POCO DIAGNOSTICADA que con frecuencia no se plantea al evaluar una hipotonía generalizada de presentación aguda. En el caso expuesto se presentan los síntomas y signos de la enfermedad descritos en la literatura especializada. Es muy importante destacar el antecedente de ingestión de alimentos ricos en carbohidratos por ser descrita como uno de los principales desencadenantes de la parálisis. El reemplazo de potasio constituye el tratamiento inicial con mejoría de la sintomatología en las horas siguientes, lo cual también ocurrió en este paciente. El hecho de no haber antecedentes de sintomatología similar en la familia no descarta el diagnóstico ya que se han encontrado una penetrancia variable de la enfermedad y en ocasiones individuos con la mutación sin ninguna sintomatología. Debido a que las crisis son fáciles de prevenir por conocerse sus desencadenantes, la mayoría de las veces debe considerarse este diagnóstico al evaluar a un niño o joven con hipotonía aguda acompañada de hipocalemia.
BIBLIOGRAFÍA
1. Bond EF. Channelopathies: potassium-related periodic paralyses and similar disorders. AACN Clin-Issues 1996; 11: 261-270. [ Links ]
2. Felix R. Channelopathies: Ion channel defects linked to heritable clinical disorders. J Med Genet 2000; 37: 729-740. [ Links ]
3. Zuberi SM, Hanna M. Ion channels and neurology. Arch Dis Child 2001; 84: 277-280. [ Links ]
4. Benartar M. Neurological potassium channelopaties. QJM 2000; 93: 787-797. [ Links ]
5. Lehmann F, Rüdel R. Channelopathies: The nondystrophic myotonies and periodic paralyses. Seminars Pediatr Neurol 1996; 3: 122-139. [ Links ]
6. Horak H, Pourmand R. Periodic paralyses. Neurol Clin 2000; 18: 195-202. [ Links ]
7. Tricarico D, Barbieri M, Camerino DC. Acetazolamide opens the muscular Kca2+ channel: a novel mechanism of action that may explain the therapeutic effect of the drug in hypokalemic periodic paralysis. Ann Neurol 2000; 48: 304-312. [ Links ]
8. Ruff RL. Insulin acts in hypokalemic periodic paralysis by reducing inward rectifier K+ current. Neurology 1999; 53: 1.556-1.563. [ Links ]
9. Morrill JA, Cannon SC. Effects of mutations causing hypokalemic periodic paralysis on the skeletal muscle L-type Ca2+ channel expressed in Xenopus laevis oocytes. J Physiol 1999; 520: 2.321-2.336. [ Links ]
10. Kim SH. Identification of mutations including de novo mutations in Korean patients with hypokalaemic periodic paralysis. Nephrol Dial Transplant 2001; 16: 939-44. [ Links ]
11. Bulman DE, Scoggan KA, Van Oene MD, Nicolle MW, Hahn AF, Tollar LL, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 1999; 53: 1.932-1.936. [ Links ]
12. Struik AF, Scoggan KA, Bulman DE, Cannon SC. The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation. J Neurosci 2000; 20: 8.610-8.617. [ Links ]
13. Jurkat RK, Lehmann-Horn F, Elbaz A, Heine R, Gregg RG, Hogan K, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA 2000; 97: 9.549-9.554. [ Links ]
14. Sternberg D, Maisonobe T, Jurkat-Rott K, Nicole S, Launay E, Chauveau D, et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 2001; 124: 1.091-1.099. [ Links ]
15. Dalakas MC, Engel WK. Treatment of permanent muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve 1983; 6: 182-186. [ Links ]
16. Tawil R, Mcdermott MP, Brown R, Shapiro BC, Ptacek LJ, Mcmanis PG, et al. Randomized trials of dichlorphenamide in the periodic paralyses. Ann Neurol 2000; 47: 46-53. [ Links ]
