










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkIatreia
versão impressa ISSN 0121-0793
Iatreia vol.16 no.1 Medellín jan./mar. 2003
PRESENTACIÓN DE CASO
Degeneración hepatocerebral: reporte de un caso pediátrico
HEPATOCEREBRAL DEGENERATION: REPORT OF A PEDIATRIC CASE
WILLIAM CORNEJO OCHOA; ANDREA RESTREPO GOUZY; JOHN EDGAR LOPERA MARÍN; JAIME CARRIZOSA MOOG
DOCTOR WILLIAM CORNEJO OCHOA, Jefe del Servicio de Neuropediatría, Universidad de Antioquia, Hospital Universitario San Vicente de Paúl, Medellín, Colombia
DOCTORA ANDREA RESTREPO GOUZY, Residente de Pediatría de la Universidad de Antioquia
DOCTOR JOHN EDGAR LOPERA MARÍN, Residente de Pediatría de la Universidad de Antioquia
DOCTOR JAIME CARRIZOSA MOOG, Profesor de Neuropediatría, Universidad de Antioquia.
LA DEGENERACIÓN HEPATOCEREBRAL ADQUIRIDA (DHCA) es un síndrome neurológico raro e irreversible que ocurre en pacientes con enfermedad hepática crónica. Se caracteriza por una disfunción progresiva de los sistemas extrapiramidal y cerebeloso. Aunque los hallazgos clínicos y de laboratorio ayudan a diferenciarla de la enfermedad de Wilson, su fisiopatología no se conoce claramente. Estos pacientes tienen hiperamonemia, pruebas anormales de tolerancia al amonio y concentraciones de manganeso elevadas. Algunos de los signos neurológicos como disartria, ataxia, temblor y demencia se presentan en el curso de la enfermedad hepática crónica con episodios recurrentes de encefalopatía hepática. Además los signos de piramidalismo están presentes en la mayoría de estos pacientes.
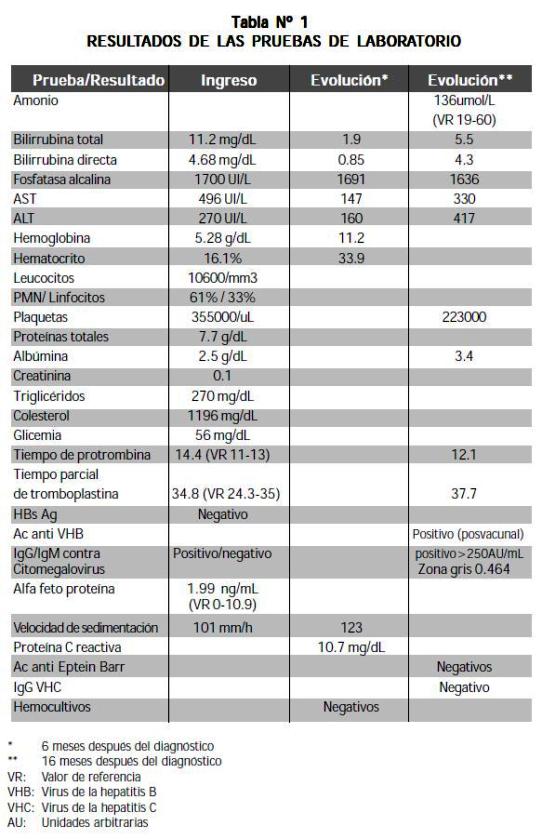
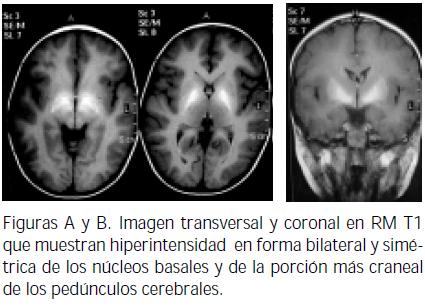
En las imágenes de T1 de resonancia magnética se observa hiperintensidad en los ganglios basales y el cerebelo aparece normal. También pueden encontrarse señales bilaterales anormales en el núcleo dentado en T2, las cuales son indistinguibles de las imágenes encontradas en la enfermedad de Wilson. Estos hallazgos en la sustancia gris pueden deberse al depósito de sustancias paramagnéticas en el putamen, globus pallidus, región subtalámica, núcleo rojo, placa cuadrigémina y porción anterior de la hipófisis.
Se describe el caso de un niño de tres y medio años con un tumor miofibroblástico hepático de un año de evolución. El examen neurológico y el estado mental del paciente eran normales, excepto por la presencia de reflejo palmomentoniano bilateral. Sin embargo, el paciente tenía los hallazgos de resonancia magnética en T1 característicos de la DHCA.
PALABRAS CLAVE
DEGENERACIÓN HEPATOCEREBRAL, ADQUIRIDA (DHCA), HEPATOPATÍA CRÓNICA, HIPERINTENSIDAD, PEDIATRÍA, RESONANCIA
SUMMARY
Acquired (non-Wilsonian) hepatocerebral degeneration (AHD) is a rare, irreversible neurologic syndrome that occurs in patients with associated chronic liver disease. It is characterized by progressive dysfunction of extrapyramidal and cerebellar systems. Although clinical and laboratory findings are helpful in the differentiation from Wilson's disease, the underlying pathophysiology has not been clearly elucidated. These patients have hyperammonemia or abnormal ammonia tolerance tests and high manganese concentrations. Some neurological signs are dysarthria, ataxia, tremor and dementia, with recurrent attacks of hepatic encephalopathy. Pyramidal tract signs are usually present.
T1-weighted images demonstrate increased signal intensity in the basal ganglia, although cerebellum seems to be spared. However, bilateral signal abnormalities in the dentate nuclei on T2-weighted images can occur in AHD, indistinguishable from those of Wilson's disease. Such gray matter lesions can be found in hepatocerebral degeneration due to deposition of paramagnetic substances in the putamen, globus pallidus, subthalamic region, red nucleus, quadrigeminal plate and anterior pituitary. We describe the case of a 3.5 year-old male patient with an hepatic miofibroblastic tumor who had been followed for one year. Neurologic examination and mental status were normal, except for the presence of bilateral palmomental reflexes and the characteristic MRI findings of AHD. There were no Kayser-Fleischer corneal rings.
In conclusion AHD can be found in children with liver disease and no apparent neurological findings and MRI abnormalities in T1 may be the only way of diagnosing this entity.
INTRODUCCIÓN
La degeneración hepatocerebral adquirida (DHCA) es una alteración heterogénea y crónica que puede suceder con una presentación primaria neurológica, hepática o combinada. Su espectro clínico es amplio y puede tener manifestaciones neuropsiquiátricas (apatía, letargia, somnolencia excesiva, convulsiones, disartria, mielopatía, agresividad, hiperactividad y deterioro de la función intelectual), motoras (ataxia, temblor, coreoatetosis, parkinsonismo, mioclonus, distonía), o ambas.
La DHCA fue descrita en 1965 por Víctor y colaboradores, quienes consideraron que era consecuencia de una exposición prolongada a las toxinas metabolizadas durante la encefalopatía hepática (1). El diagnóstico es difícil de hacer en los estadios iniciales porque los pacientes pueden ser asintomáticos neurológicamente. Las imágenes de resonancia magnética (RNM) han dado una luz en el conocimiento de esta entidad, describiendo una hiperintensidad en T1 en el globus pallidus, putamen, mesencéfalo, caudado y cápsula interna principalmente. Estos hallazgos se han descrito en pacientes con cirrosis alcohólica, cirrosis biliar primaria, uso prolongado de nutrición parenteral total (NPT), colestasis, hemocromatosis, hepatitis autoinmune y en aquéllos a quienes se les ha realizado derivación portosistémica (2-12). La fisiopatología de esta lesión aún se desconoce. La mayoría de los casos se han descrito en adultos, en los cuales la literatura sugiere que el comportamiento es progresivo e irreversible (13). Los casos descritos en la población infantil son escasos.
CASO CLÍNICO
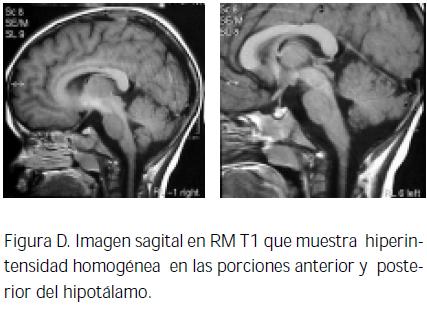
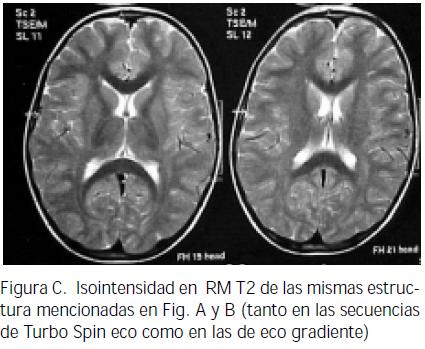
Paciente de sexo masculino de 3 años y medio, producto del primer embarazo, parto espontáneo en vértice. Consultó a la edad de 26 meses por 3 meses de evolución de adinamia, hiporexia, prurito, ictericia, orinas hiperpigmentadas y acolia, además fiebre de intensidad no determinada. Dos meses antes había presentado hematoquezia, con resolución espontánea. No tenía antecedentes personales de ictericia al nacimiento. Su esquema de vacunación estaba completo para la edad, no había recibido transfusiones ni había tenido hepatitis. No tenía antecedentes familiares de hepatitis. Al examen físico estaba eutrófico, peso, 14.800 gramos, frecuencia cardíaca, 98 por minuto, temperatura, 37.5° C. Los hallazgos positivos fueron: ictericia generalizada, adenopatías de 1 cm, bilaterales en cuello y axilas, móviles, indoloras, abdomen globuloso, doloroso a la palpación, hepatomegalia de 8 centímetros debajo del reborde costal derecho, examen neurológico normal. No se encontró anillo de Kayser-Fleischer. Recibió ampicilina sulbactam, fenobarbital, difenhidramina, albúmina, furosemida, ácido ursodeoxicólico y analgesia; no requirió nutrición parenteral. Las pruebas de laboratorio aparecen en la tabla N° 1. La ecografía abdominal demostró una dilatación marcada de la vía biliar intrahepática, con lesión nodular, hipodensa de 20x24 mm en la confluencia de los conductos hepáticos. La colangiorresonancia mostró una lesión de 2 cm de diámetro en la concurrencia de los conductos hepáticos, con dilatación de la vía biliar intrahepática. Se hicieron laparotomía diagnóstica y biopsia de hígado y ganglios hiliares; se huicieronontró una masa dura y amarilla de 2 cm de diámetro, localizada en el conducto hepático común. El análisis histológico de los ganglios evidenció una hiperplasia folicular con linfadenitis crónica, sin malignidad. La vesícula biliar presentaba inflamación crónica. En el hígado se encontró un tumor miofibroblástico inflamatorio de la porta hepatis. Un mes después se efectuaron resección del área fibrótica, portoenteroanastomosis y colecistectomía. La gamagrafía hepatobiliar fue normal. Nuevamente fue hospitalizado a los 34 meses de edad por dolor en hemiabdomen superior y fiebre de 40° C. Al examen físico se encontraron ictericia sin acolia, hepatomegalia de 4 cm por debajo del reborde costal derecho y examen neurológico normal. En la tomografía simple y contrastada de abdomen persistían pequeñas áreas de alteración periportal correspondientes a una lesión residual o recidivante, con mínima dilatación biliar intrahepática. La ecografía abdominal mostró el hígado aumentado de tamaño, con un diámetro mayor de 12.2 centímetros en el lóbulo derecho y de 7.5 cm. en el izquierdo, sin dilatación de la vía biliar intrahepática, ni líquido en cavidad; no había adenopatías ni masas intrabdominales; el bazo se encontraba aumentado de tamaño (8.4 cm). El último ingreso tuvo lugar a la edad de tres y medio años, al servicio de urgencias, por 1 día de evolución de tos disfónica y estridor, compatible con laringotraqueítis viral, lo cual le produjo un episodio de sofocación, con cianosis y convulsión secundaria a la hipoxia. Al examen físico se hallaron telangiectasias e hirsutismo en cara y múltiples adenopatías de más de 1 cm de diámetro en el cuello y las axilas. El hígado estaba indurado, a 9 cm del reborde costal derecho, sin ascitis. Debido a los estigmas de hepatopatía crónica, con pruebas de coagulación alteradas y la asociación del episodio convulsivo, se solicitó estudio imaginológico. La tomografía simple de cráneo no evidenció hallazgos patológicos. En la secuencia T1 de la resonancia magnética se encontró hiperintensidad bilateral y simétrica de los núcleos basales, de la porción superior de los pedúnculos cerebrales (figuras A y B) y de las porciones anterior y posterior de la hipófisis (Figura D). Había isointensidad en T2, tanto en las secuencias de Turbo Spin eco como en las de eco gradiente (figura C); no había efecto de masa, edema perilesional ni realce postcontraste. Tampoco se encontraron infarto, malformación vascular ni lesión inflamatoria o infecciosa, intra o extraaxial. Los hallazgos imaginológicos orientaban hacia la existencia de una degeneración hepatocerebral adquirida por hepatopatía crónica. La colangiorresonancia mostró signos de lesión neoplásica recidivante, con obstrucción de la vía biliar en la confluencia de los conductos hepáticos. Por ello se llevó a cirugía y se encontraron fibrosis hepática, hiperplasia sinusal de los ganglios linfáticos y cirrosis biliar .
El examen neurológico mostró que era un niño alerta, irritable por momentos, pero con un estado mental intacto, marcha normal, simetría facial, pares craneales normales, fondo de ojo normal, sin paresias, reflejos osteotendinosos normales, respuesta plantar flexora bilateral, fuerza muscular conservada, pruebas de coordinación normales y sin alteraciones práxicas; el examen sensitivo fue normal. El único hallazgo positivo era un reflejo palmomentoniano bilateral (Marinesco)
DISCUSIÓN
Se reporta el caso de un niño de 3 años y medio con cirrosis biliar secundaria a un tumor miofibroblástico inflamatorio de la porta hepatis, a quien se le realizó una portoenteroanastomosis, a pesar de la cual persistió la colestasis. Al evaluarlo llamó la atención que el examen neurológico era prácticamente normal excepto por un signo palmomentoniano bilateral. La ausencia de signos neurológicos en este caso, contrasta con lo reportado en la literatura, en la cual la mayoría de los pacientes son adultos y presentan alguna alteración neurológica, tal como deterioro cognitivo, aumento de los reflejos osteotendinosos, Babinski, dificultad para la marcha, hemiparesia, temblor intencional, disartria, movimientos coreiformes de la lengua, habla hipotónica (1,2,13). Sinan et al (13) reportaron en un paciente de 54 años con cirrosis postnecrónica los hallazgos anteriormente mencionados, asociados a la presencia del reflejo palmomentoniano, al igual que en nuestro paciente. Otros autores, como Uchino A. et al., (6) reportaron 4 pacientes con cirrosis biliar primaria y degeneración hepatocerebral subclínica. La mayoría de los casos han sido descritos en adultos con enfermedad hepática crónica y episodios de encefalopatía hepática repetidos y que por lo tanto han tenido un examen neurológico anormal; pensamos que nuestro paciente es aún asintomático por la corta evolución de la hepatopatía.
En la literatura se describe en forma constante una imagen hiperintensa, simétrica y bilateral en la RM T1 en el globus pallidus, putamen y núcleo caudado y de forma variable en el mesencéfalo, lámina cuadrigémina, cápsula interna, región subtalámica, núcleos rojos, cerebelo, porción media e hipófisis (1-3,6,14-16). Es importante recordar que las causas de imagen hiperintensa en la RM T1 son: la grasa, el sangrado subagudo y el melanoma. Las imágenes en la RM T2 se describen como hipointensas en la mayoría de los estudios, sin embargo, algunos autores como Genovese et al. (14) reportan imágenes hiperintensas. Las causas y mecanismos del incremento en la intensidad de la señal son aún desconocidas. Sin embargo, existen varias hipótesis: la asociación entre señales hiperintensas en el cerebro y derivaciones portosistémicas sugiere que el depósito de sustancias paramagnéticas puede jugar un papel en la patogénesis. Actualmente la hipótesis más aceptada de la causa de la hiperintensidad en la RM T1 en los núcleos de la base son los depósitos de manganeso (Mn), un metal de transición paramagnética (17-20). En cuanto a la captación en la RM T2 son necesarias concentraciones extremadamente elevadas de Mn, incompatibles con la vida (21). Newland et al. (17) ha descrito hiperintensidad de la señal en la RM T1 de los núcleos de la base y de la hipófisis, luego de la administración parenteral de 5 mg/kg de clorhidrato de manganeso en macacos. Krieger et al. (18) correlacionan estos mismos hallazgos con los niveles sanguíneos de Mn de los pacientes con enfermedad hepática terminal. En personas saludables no existen depósitos de Mn en el sistema nervioso central (SNC), debido a un mecanismo de autorregulación entre el sistema gastrointestinal y el hígado (20). Normalmente, del 1.0 al 3.5% de la ingestión oral de Mn es absorbido por la circulación sistémica; el 98% del mismo es metabolizado por el hígado y eliminado por la vía biliar (22,23). Sin embargo, la presencia de circulación colateral portosistémica puede desviar el metabolismo del Mn y hacer que se deposite en el SNC (24). Su paso a través de la barrera hematoencefálica es mediado por la transferrina. El efecto tóxico sobre el tejido nervioso se debe al Mn+3 libre e inestable con acción oxidante sobre la dopamina y autoxidación en neuromelanina (25). El acúmulo de la neuromelanina puede ser también responsable de la hiperintensidad en la RM T1 de los núcleos de la base (26).
Es importante considerar que más de la mitad de los niños con colestasis por uso prolongado de NPT, que contiene manganeso, tienen imágenes anormales en los ganglios basales (8,14). En cuanto al amonio, no es considerado como una sustancia paramagnética. Su acumulación por descenso de la conversión del amonio a úrea en la insuficiencia hepática determina la aparición de una hiperintensidad en la RM T1, la cual ocurre sólo en concentraciones elevadas (27-29). Los altos niveles de amonio van a producir daños en los astrocitos, que son vulnerables a la toxicidad por esta sustancia, lo cual provoca lesión de la barrera hematoencefálica y acumulación de líquido y de macromoléculas, causando una disfunción de la membrana y de las organelas del astrocito. Estos cambios se traducen en hiperintensidad en RM T1 (1).
En la variante Huttenlocher del síndrome de Alpert se ha descrito degeneración hepatocerebral de comienzo en la niñez. Estos pacientes pueden presentar convulsiones parciales secundariamente generalizadas, estatus epiléptico, deterioro psicomotor, y disfunción hepática que se puede exacerbar con la administración de ácido valproico (30).
Algunos pacientes con hemocromatosis presentan signos neurológicos similares a los de los pacientes con degeneración hepatocerebral, pero al contrario de estos últimos, en los primeros hay una hiperintensidad en RM T2. Es aún incierto el papel de las cargas anormales de hierro en la explicación de estos hallazgos (11).
La histopatología en los pacientes adultos con degeneración hepatocerebral muestra necrosis laminar cortical y polimicrocavitaciones en la corteza y los ganglios basales, a lo cual se asocian atrofia cerebral y cerebelosa, mielinolisis póntica central y extrapóntica en los cuerpos geniculados, tálamo, cápsula interna, fórnix, tracto mamilotalámico, núcleo caudado, pallidum, núcleo oculomotor y sustancia blanca cerebelosa (15,31). Microscópicamente es característico ver astrocitos tipo II de Alzheimer y gránulos citoplasmáticos de glucógeno (32). Los hallazgos en la RM ubican estas lesiones en forma simétrica y bilateral en la sustancia blanca de los hemisferios cerebrales. Sin embargo, las imágenes anormalmente hiperintensas observadas en el núcleo dentado sugieren que la sustancia gris también está implicada, como se ha reportado en estudios previos (33,34).
Las consecuencias neurológicas de estos hallazgos en la resonancia en niños aún no están claras. Algunos estudios sugieren reversibilidad de las lesiones al corregir la función hepática (35).
Existen reportes acerca de la desaparición de las alteraciones de la señal después del trasplante hepático, de la suspensión del Mn en la dieta parenteral, y luego de la embolización terapéutica de la derivación venosa porto-sistémica intrahepática (36-38).
Luttmann et al. (38), en 1997 describieron el caso de un paciente con encefalopatía por VIH e hiperintensidad en RM T1 en la sustancia nigra y los tractos corticopónticos, bilaterales y simétricos.
Se puede concluir que la evaluación neurológica, apoyada por un estudio de neuroimágenes, especialmente la RM T1, es fundamental al estudiar clínicamente un paciente con enfermedad hepatobiliar, para diagnosticar en forma temprana la degeneración hepatocerebral, principalmente en la niñez, ya que su sintomatología y signología neurológica son muy escasas.
AGRADECIMIENTOS
Al Doctor Jorge Holguín, profesor emérito de la Universidad de Antioquia, por la revisión del presente manuscrito.
BIBLIOGRAFÍA
1. VICTOR M, ADAMS RD, COLE M. The Adquired (nonwilsonian) type of chronic hepatocerebral degeneration. Medicine 1965; 44: 345-396. [ Links ]
2. VICTOR M. Persistent altered mentation due to ethanol. Neurol Clin 1993; 11: 639-661. [ Links ]
3. CHARNESS ME. Brain lesions in alcoholics. Alcohol Clin Exp Res 1993; 17: 2-11. [ Links ]
4. VICTOR M. Alcoholic dementia. Can J Neurol Sci 1994; 21: 88-99. [ Links ]
5. KRIL JJ, BUTTERWORTH RF. Diencephalic and cerebellar pathology in alcoholic and nonalcoholic patients with end-stage liver disease. Hepatology 1997; 26: 837-841. [ Links ]
6. UCHINO A, HASUO K, MATSUMOTO S, MASUDA K. Cerebral MR imaging in patients with primary biliary cirrhosis. Nippo Igaku Hoshasen Gakkai Zasshi 1993; 53: 145-149. [ Links ]
7. BLEASEL AF, WAUGH RC, MCCAUGHAN GW. Development of chronic hepatocerebral degeneration eight years after a distal splenorenal (Warren) shunt. Gut 1989; 30: 1.419-1.423. [ Links ]
8. MIROWITZ SA, WESTRICH TJ, HIRSCH JD. Hyperintense basal ganglia on T1-weigted MR images in patients receiving parenteral nutrition. Radiology 1991; 179: 551-555. [ Links ]
9. MAGHNIE M, GENOVESE E, MAGGIORE G. Central nervous system hyperintensity on magnetic resonance imaging in children with cholestatic liver disease. Eur J Pediatr 1995; 154: 938-942. [ Links ]
10. WERNECK AL, GDALEVICI C, FONSECA RI. Neuroimagem na degeneracao hepatocerebral induzida por hemocromatose. Arq Neuropsiquiatr 1997; 55: 625-631. [ Links ]
11. DEMARQUAY G, SETIEY A, MOREL Y, TREPO C, CHAZOT G, BROUSSOLLE E. Clinical report of three patients with hereditary hemochromatosis and movement disorders. Mov Disord 2000; 15: 1.204- 1.209. [ Links ]
12. SPENCER DC, FORNO LS. Dementia with motor dysfunction in a patient with liver disease. Brain Pathol 2000; 10: 315-319. [ Links ]
13. SINAN L, KURUOGLU R, BAYSAL AI, IRKEC C, AKKUS-ASLAN S. Acquired hepatocerebral degeneration: a case presentation with cranial magnetic resonance imaging findings. Gazi Med J 1998; 9:136-138. [ Links ]
14. GENOVESE E, MAGHNIE M, MAGGIORE G, TINELLI C, LIZZOLI F, DE GIACOMO C, et al. MR imaging of CNS involvement in children affected by chronic liver disease. Am J Neuroradiol 2000; 21: 845-851. [ Links ]
15. JOG MS, LANG AE. Chronic acquired hepatocerebral degeneration: case reports and new insights. Mov Disord 1995; 10: 714-722. [ Links ]
16. CONDAT B, DUSOLEIL A, BERNARDEAU M, ROCHE A, PELLETIER G, BUFFET C. Degenerescence hepatocerebrale chronique acquise: role du manganese et traitement par occlusion endovasculaire d'un shunt porto- systemique. Gastroenterol Clin Biol 1999 ; 23: 268-270. [ Links ]
17. NEWLAN MC, CECKLER TL, KORDOWER JH, WEISS B. Visualizing manganese in the primate basal ganglia with MRI. Exp Neurol 1989; 106: 251 258. [ Links ]
18. KRIEGER D, KRIEGER S, JANSEN O, GASS P, THEILMANN L, LIEHTNECKERH. Manganese and chronic hepatic encephalophaty. Lancet 1955; 346: 270-274. [ Links ]
19. MIROWITZ AS, WESTRICH TJ. Basal ganglia signal intensity alterations: reversal after discontinuation of parenteral manganese administration. Radiology 1992; 185: 535-536. [ Links ]
20. MARKESBERY WR, EHMANN WD, HOSSAIN TI, ALAUDDIN M. Brain manganese concentrations in human aging and Alzheimer's disease. Neurotoxicology 1984; 5: 49-58. [ Links ]
21. NELSON K, GOLNICK J, KORN T, ANGLE C. Manganese encephalopathy: utility of early magnetic resonance imaging. Br J Ind Med 1993; 50: 510-513. [ Links ]
22. GREENBERB DM, COPP DH, CUTHBERTSON EM. Studies in mineral metabolism with aid of artificial radioactive isotopes; distribution and excretion particulary by way of bile, of iron, cobalt and manganese. J Biol Chem 1943: 147: 749-756. [ Links ]
23. LARSEN NA, PAKKENBERG H, DAMSGAARD E, HEYDORN K. Topographical distribution of arsenic, manganese, and selenium in the normal human brain. J Neurol Sci 1979; 42: 407-416. [ Links ]
24. MATSUMOTO S, MORI H, YOSHIOKA K, KIYOSUE H, KOMATSU E.Effects of portal systemic shunt embolization on the basal ganglia: MRI. Neuroradiology 1997; 39: 326-328. [ Links ]
25. DONALDSON J, LA BELLA FS, GESSER D. Enhanced autoxidation of dopamine as a possible basis of manganese neurotoxicity. Neurotoxicology 1981; 2: 53-64. [ Links ]
26. GOZZOLI L, AMBROGIO L, GRASSO M. Iperintensita dei nuclei della base nelle sequenze RM T1-W in un caso di shunt porto-sistemico senza insufficienza epatica. Revista di Neuroradiologia 1996; 9: 493-500. [ Links ]
27. KULISEVSKI J, PUJOL J, GRAU JM. Pallidal hyperintensity on MRI in cirrhotic patients: clinical correlations. Hepatology 1992; 16:1.382-1.388. [ Links ]
28. KULISEVSKI J, PUJOL J, JUNQUE C, DEUS J, BALANZO J, CAPDEVILA A. MRI pallidal hyperintensity and brain atrophy in cirrhotic patients: two different MRI patterns of clinical deterioration. Neurology 1993; 43: 2.570-2.573. [ Links ]
29. VYMAZAL J, BABIS M. T1 and T2 alterations in the brains of patients with hepatic cirrhosis. Am J Neuroradiol 1996;17: 333-336. [ Links ]
30. BICKNESE AR, MAY W, HICKEY WF, DODSON WE. Early childhood hepatocerebral degeneration misdiagnosed as valproate hepatotoxicity. Ann Neurol 1992; 32: 776-775. [ Links ]
31. SOFFER D, SHERMAN Y, TUR-KASPA R, EID A. Acquired hepatocerebral degeneration in a liver transplant recipient. Acta Neuropathol 1995; 90: 107- 111. [ Links ]
32. LEE J, LACOMIS D, COMU S, JACOBSOHN J, KANAL E. Acquired hepatocerebral degeneration: MR and pathologic findings. Am J Neurorad 1998; 19: 485-487. [ Links ]
33. BRUNBERG JA, KANAL E, HIRSCH W, VAN THIEL DH. Chronic acquiered hepatic failure: MR imaging of the brain at 1.5 T. Am J Neurorad 1991; 2: 909-914. [ Links ]
34. HANNER JS, LI KC, DAVIS GL. Acquired hepatocerebral degeneration: MR similarity with Wilson Disease. J Comput Assist Tomogr 1988; 12: 1.076-1.077. [ Links ]
35. BALLAUFF A, ENGELBRECHT V, VOIT T. Hyperintense lesions of the globus palidus on MRI in children with chronic liver disease. Eur J Pediatr 94; 153: 802-806. [ Links ]
36. KULISEVSKY J, PUJOL J, DEUS J, JUNQUE C, BALANZO J, AVILA A, CAPDEVILA A. Persistence of MRI hyperintensity of globus pallidus in cirrhotic patients: a 2 year follow-up study. Neurology 1995; 45: 995-997. [ Links ]
37. MIROWITZ SA, WESTRICH TJ, HIRSH JD. Basal ganglia signal intensity alterations: reversal after discontinuation of parenteral manganese administration. Radiology 1992; 185: 535-536. [ Links ]
38. LUTTMANN S, HUSSTEDT IW, SCHUIERE. Highsignal lesions in the midbrain on T1-weighted MRI in an HIV-infected patient. Neuroradiology 1997; 9: 36-138. [ Links ]
