










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.16 supl.1 Medellín Mar. 2003
Evaluación de la mutación a3243g en mtDNA, en familias de pacientes diagnosticados con el síndrome Melas
Victoria Parra1; Carlos Burgos2; William Cornejo2; Gabriel Bedoya3; Andrés Ruiz4
1 Estudiante de Maestría, Posgrado en Ciencias Básicas Biomédicas. dqvicky@yahoo.com
2 Profesor, Facultad de Medicina, Universidad de Antioquia
3 Profesor, Facultad de Ciencias Exactas y Naturales, Universidad de Antioquia
4 Profesor, Facultad de Medicina, Universidad de Antioquia; Profesor Universidad de Londres
INTRODUCCIÓN Y OBJETIVOS
Las citopatías mitocondriales constituyen un variado grupo de desórdenes generados por déficits de la producción de energía en la mitocondria (1), proceso llevado a cabo a través de cinco complejos multienzimáticos ubicados en la membrana interna mitocondrial. Las subunidades que conforman estos complejos son codificadas por genoma nuclear y mitocondrial (mtDNA). Hasta el momento se ha identificado un gran cantidad de citopatías causadas por mutaciones en mtDNA; la más frecuente es MELAS (Mitochondrial Encephalomyopathy with Lactic acidosis and Stroke-like episodes) (2), de ésta, el 80% de los casos poseen la mutación A3243G en el gen del tRNALeu (3). En dicha mutación se ha encontrado hasta un 95% de heteroplasmia (4), lo cual hace que la variación en el fenotipo sea muy amplia.
En este trabajo se evaluó la mutación A3243G en pacientes con diagnóstico de MELAS así como a sus familiares.
METODOLOGÍA
Se obtuvo ADN de diferentes tejidos (sangre, músculo y/o carrillo bucal) de 60 pacientes con sospecha de MELAS. Se amplificó por PCR un fragmento de 533 pb del genoma mitocondrial que incluía el gen para el tRNALeu y parte del gen ND1. Los amplificados fueron purificados y posteriormente secuenciados. En los pacientes quienes se detectó una mutación, se procedió a evaluar clínica y genéticamente a familiares por línea materna. La genotipificación se realizó mediante la técnica PCR-RFLPs utilizando la enzima Apa I, cuyo sitio de reconocimiento es generado por la mutación.
RESULTADOS Y CONCLUSIONES
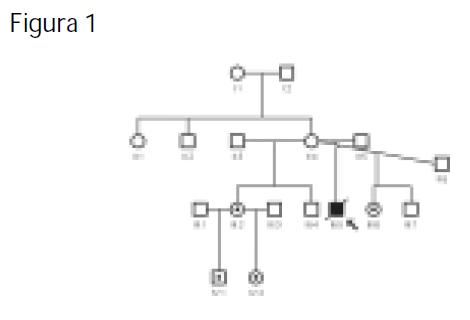
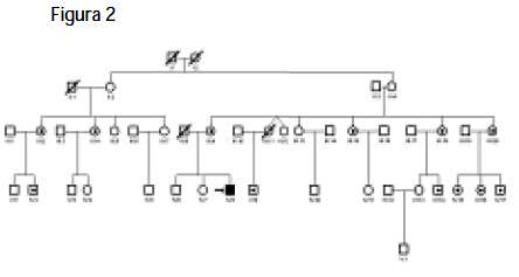
De un total de 60 pacientes con sospecha de MELAS, sólo en dos se detectó la mutación A3243G. Con la evaluación de familiares de estos pacientes (Figuras 1 y 2), encontramos que otros miembros de las familias eran portadores de la mutación A3243G pero no tenían el cuadro clínico de MELAS, con lo que se logró asociar esta transición a diferentes entidades como: hipoacusia, migraña, talla baja y posiblemente diabetes mellitus tipo 2 (una integrante de una de las familias es diabética y presenta la mutación). Además, se observa una clara correlación entre la cantidad de mtDNA mutado y la severidad de los síntomas.
PALABRAS CLAVE
mtDNA, MELAS, A3243G, MUTACIÓN MITOCONDRIAL, HETEROPLASMIA
BIBLIOGRAFÍA
1. RUBIO JC, et al (1998) Déficits de los complejos enzimáticos de la cadena respiratoria mitocondrial. Revista de Neurología 1998; 26 (Supl 1): S15-S20. [ Links ]
2. PAVLAKIS SG et al (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like epiosodes (MELAS). Annals 1984; 16: 481-488. [ Links ]
3. GOTO YI, NONAKA I, HORAI S (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990; 13: 651-653. 4. Mitosyn Database: www.neuro.wustl.edu/neuromuscular/mitosyn.html [ Links ]
