










Serviços Personalizados
Journal
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkIatreia
versão impressa ISSN 0121-0793
Iatreia v.17 n.2 Medellín abr./jun. 2004
ARTÍCULO DE REVISIÓN
Mutaciones de los canales neuronales de sodio y cloro asociadas a epilepsia generalizada con convulsiones febriles plus
MUTATION OF NEURONAL CHANNELS OF SODIUM AND CHLORIDE ASSOCIATED WITH GENERALIZED EPILEPSY WITH FEBRILE SEIZURES PLUS (GEFS+)
DIANA CATALINA ALZATE MONSALVE1; JAIME CARRIZOSA MOOG2; GABRIEL BEDOYA BERRÍO3
1 Laboratorio de Genética Molecular.
2 Profesor del Departamento de Pediatría y Puericultura Universidad de Antioquia.
3 Laboratorio de Genética Molecular. Universidad de Antioquia, Facultad de Medicina, Medellín-Colombia.
RESUMEN
LA EPILEPSIA GENERALIZADA CON CONVULSIONES FEBRILES PLUS (EGCF+), es una entidad relativamente común. Se caracteriza por convulsiones de tipo generalizado con una gran variabilidad fenotípica; se presenta desde los 3 meses de edad y persiste más allá de los 6 años; las convulsiones pueden ser precipitadas por fiebre pero se presentan también sin ella. La enfermedad se ha asociado a herencia autosómica dominante con penetrancia incompleta, en la que intervienen mutaciones de los genes que codifican los canales iónicos de sodio dependientes del voltaje y de los canales iónicos de cloro en las neuronas del Sistema Nervioso Central (SNC). El amplio fenotipo de la EGCF+ se ha encontrado en asociación con otras entidades como la Epilepsia Mioclónica Severa del Lactante (EMSL) y la Epilepsia Generalizada Tónico-Clónica Intratable de la Infancia (EGTCII), las cuales han presentado mutaciones comunes con las de la EGCF+, según informes recientemente publicados. Esta revisión pretende recopilar información de la literatura publicada sobre la EGCF+, con el objeto de brindar al lector un mejor conocimiento de esta entidad y de su asociación con las mutaciones que participan en su patogenia.
PALABRAS CLAVE
EPILEPSIA GENERALIZADA CON CONVULSIONES FEBRILES PLUS RECEPTORES CELULARES, CANALOPATÍAS, MUTACIÓN.
SUMMARY
Generalized Epilepsy with Febrile Seizures Plus (GEFS+) is a frequent entity characterized by generalized seizures with a wide phenotypic variety; the age of onset is 3 months and it persists beyond 6 years. Seizures may or may not be induced by fever. The disease has shown an autosomic dominant trait, incomplete penetrance and association with mutations on the genes that encode voltage-dependent sodium channels and the chloride neuronal channels on the central nervous system. The wide spectrum GEFS+ phenotype has been related with others entities such as Severe Myoclonic Epilepsy of Infancy (SMEI) and Intractable Childhood Epilepsy with Frequent Generalized Tonic-Clonic Seizures (ICEGTC); they have mutations in common with GEFS+ according to several recently published articles. This review compiles up to date information about EGCF+ with the aim of giving the reader a knowledge of this entity and of its association with mutations that participate in its pathogenesis.
KEY WORDS
GENERALIZED EPILEPSY WITH FEBRILE SEIZURES PLUS (GEFST), CELL RECEPTORS, CANALS MUTATIONS, CANALS PATHOLOGY.
INTRODUCCIÓN
LAS FUNCIONES CORPORALES que se realizan en todo momento están estrictamente reguladas por una comunicación continua y coordinada entre los grupos celulares de los diferentes tejidos, lo que corresponde a una compleja red de intercambio efectuado mediante señales que utilizan como mediadores los neurotransmisores y las hormonas, entre otros, con la presencia necesaria de receptores en las células que efectúen las órdenes correspondientes. Las alteraciones de esta comunicación pueden tener diversas manifestaciones clínicas; entre las que afectan al funcionamiento de los receptores celulares se encuentra el grupo de las epilepsias. A continuación se hará una descripción de los receptores celulares importantes en la patogenia de las epilepsias y se detallarán las mutaciones publicadas de los receptores neuronales asociadas a la Epilepsia Generalizada con Convulsiones Febriles Plus (EGCF+).
TIPOS DE RECEPTORES CELULARES IMPORTANTES EN LA PATOGENIA DE LAS EPILEPSIAS
LOS CANALES IÓNICOS juegan un papel importante en la etiología de los síndromes epilépticos, pues participan en la generación y mediación de señales celulares en las membranas excitables respondiendo a los cambios del potencial de membrana, a los ligandos extracelulares o a segundos mensajeros. Se han identificado los siguientes:
1. Canales dependientes del voltaje como los de Ca+, Na+, K+.
EL CANAL DE CA+ dependiente del voltaje participa en la liberación de neurotransmisores a partir de las neuronas, lo cual se lleva a cabo a través de la despolarización celular en la que aumenta el contenido de Ca++ en el interior de la célula con disminución de este electrólito en el espacio extracelular.
El canal de Na+ está conformado por una subunidad a y dos subunidades β. La subunidad a puede funcionar por sí sola en la despolarización celular, pero se necesitan las subunidades β para regular el momento de cierre del canal. Se han implicado mutaciones en ambos tipos de subunidades en la patogenia de la EGCF+.
El canal iónico de K+ participa en la repolarización e hiperpolarización de las membranas celulares; se encuentra conformado por 4 subunidades; la mutación de una ellas puede interferir en el control que este canal hace en la excitación celular (1).
2. Canales iónicos relacionados con ligandos extracelulares: colinérgico nicotínico, gabaérgico y glutaminérgico.
EL CANAL COLINÉRGICO NICOTÍNICO tiene la capacidad de generar un potencial de membrana que cuando es lo suficientemente alto produce apertura de los canales de sodio dependientes del voltaje. Estos que reconoce su ligando, una parte transmembrana y un componente intracelular que se une a segundos mensajeros. El canal está conformado por 5 subunidades que pueden ser α, β, γ, o, y δ, con múltiples variables para las subunidades α y β, las que combinadas de diferentes maneras confieren variabilidad a la configuración del canal.
Los canales gabaérgicos pueden ser de dos tipos; el A, llamado receptor de cloro, está conformado por 5 subunidades, 2 α, 2 β y 1 γ; este tipo es dependiente del voltaje y su mutación también ha sido implicada en el síndrome de EGCF+. El tipo B se ha denominado receptor de potasio, está conformado por dos subunidades acopladas a las proteínas G intracelulares, las cuales participan como segundos mensajeros.
Los canales glutaminérgicos son de dos tipos: los metabotrópicos, que son activados por segundos mensajeros, y los ionotrópicos que son dependientes del voltaje. Entre estos últimos están los receptores activados por N-metil-D-aspartato y por glutamato (NMDA), y los receptores no NMDA; éstos incluyen los receptores AMPA (activados por aminohidroxi- 5metil-isoxazol-propionil y por glutamato) y los receptores KA (activados por kainato, domoato y glutamato) (1).
Se describirán a continuación los canales asociados a la patogenia de la EGCF+; como ya se mencionó, ellos son los canales de sodio dependientes del voltaje y los canales gabaérgicos tipo A (canal de cloro).
a. Canal de sodio dependiente del voltaje
Está conformado por tres subunidades: α , β1, β2.
La subunidad α tiene 4 dominios que conforman el poro de Na+. Cada dominio está formado por 6 segmentos transmembrana, el cuarto de los cuales actúa como sensor de voltaje y los segmentos quinto y sexto de los cuatro dominios forman el poro del canal. La subunidad β1 de los canales de sodio dependientes del voltaje es una proteína integral de membrana que tiene una región simple transmembrana y un dominio amino-terminal extracelular prominente (2) (Figura N° 1).
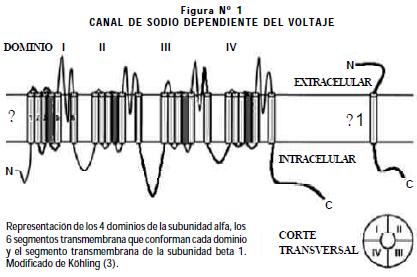
Los diferentes tipos de canales pueden distinguirse con respecto a sus subunidades a, varias de las cuales han sido clonadas. En el hombre, las subunidades a son codificadas por un mínimo de cinco genes diferentes; entre estos se encuentran SCN1A, SCN2A, SCN3A, SCN5A, SCN6A y SCN8A (3).
Las subunidades α1 se encuentran codificadas en el gen SCN1A y las α2 en el gen SCN2A, ambas del locus 2q24 (1). Las subunidades β1 se encuentran codificadas en el gen SCN1B del locus 19q13.1 (1). Estas subunidades β no son esenciales para la función del canal pero pueden acelerar su activación e inactivación (3).
Las mutaciones de los locus de las subunidades α1, α2 y β1 se han asociado a EGCF+.
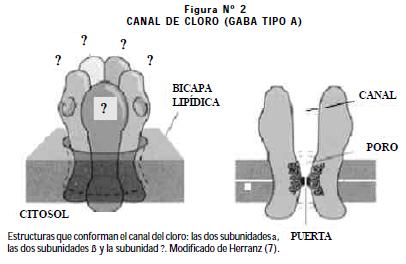
b. Canal iónico de GABA tipo A (canal de cloro)
Está formado por 5 subunidades: dos alfa, dos beta y una gamma, cada una con 4 dominios transmembrana (M1, M2, M3 y M4); la unión de los dominios M2 de las 5 subunidades forma el poro del canal de Cl- (1). Las subunidades de este canal, al igual que las del canal de sodio, se numeran de acuerdo con el gen que las codifica; un ejemplo de esto es la mutación de la subunidad γ2 localizada en el gen GABRG2 del locus 5q que se ha asociado a la EGCF+ (Figura N° 2).
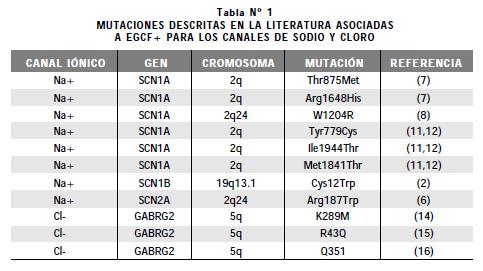
En la tabla N° 1 pueden observarse las mutaciones de los canales de sodio y cloro,asociadas a la EGCF+, que han sido descritas hasta el momento.
CARACTERIZACIÓN CLÍNICA DE LA EGCF+
LA EGCF+ fue descrita por primera vez en 1997 por Scheffer y Berkovic (4) quienes la identificaron mediante el estudio de una familia con 25 personas afectadas en 4 generaciones. El fenotipo de la entidad se caracteriza por el inicio precoz (a partir de los 3 meses de edad) de convulsiones precipitadas por la fiebre, las cuales persisten más allá de los 6 años (convulsiones febriles plus CF+) pero que pueden presentarse también como crisis convulsivas afebriles.
Las CF+ se diferencian de las convulsiones febriles simples (CFS) porque en las primeras los ataques con la fiebre continúan después de los 6 años o se asocian a convulsiones tónico-clónicas afebriles, con fenotipos menos comunes que incluyen ausencias, convulsiones mioclónicas o atónicas (4); en contraste, las CFS nunca se presentan después de los 5 años de edad y tienen una prevalencia mayor en la población infantil (3-5%).
La severidad de las convulsiones en el síndrome EGCF+ es muy variable, lo cual no está relacionado con factores adquiridos, como trauma o infección cerebral. Esta variabilidad puede explicarse porque la expresión fenotípica depende del sitio del canal donde ocurre la mutación. La expresión de esta, en puntos que son claves en la estructura y funcionamiento del canal del Na como los receptores o los poros, produce convulsiones más severas que si se localizan en sitios menos relevantes para el funcionamiento del canal.
El síndrome EGCF+ se expresa en los individuos de una manera autosómica dominante con un 60% de penetrancia; el diagnóstico se establece cuando se encuentran los siguientes elementos clínicos:
- Ausencia de factores adquiridos que predispongan a epilepsia.
- Historia familiar de fenotipos similares.
- Convulsiones precipitadas por la fiebre y/o convulsiones afebriles.
- Fenotipo característico de convulsiones desde los 3 meses de vida hasta más allá de los 6 años de edad, como única manifestación clínica (5).
El pronóstico del síndrome EGCF+ es más reservado que el de las CFS.
MUTACIONES ASOCIADAS A LA EGCF+
El fenotipo de las CF+ se ha encontrado asociado a la mutación de los genes que codifican los canales de sodio dependientes del voltaje, los cuales están formados, como ya se dijo, por una subunidad α y dos subunidades β (β1y β2), al igual que con la mutación de la subunidad γ2 del canal del cloro de GABA.
Mutaciones del canal de sodio dependiente del voltaje
LAS SUBUNIDADES α1 y α2 presentan diferentes distribuciones en las neuronas y también en varias regiones del cerebro: la primera está localizada en el cuerpo y la segunda en los axones de las neuronas, lo que puede conducir a diferencias en el fenotipo de las convulsiones (6).
En las publicaciones hechas por los grupos de Escayg y Wallace (7-10) la mutación de SCN1B en 19q13.1 se relaciona con la EGCF+ tipo 1 y las mutaciones de SCN1A y SCN2A en 2q24 condicionan la de tipo 2. Los estudios de ligamiento indican que la prevalencia de EGCF+ tipo 2 es mayor que la del tipo 1 (9). Aunque se ha clasificado la EGCF+ dependiendo de que la mutación afecte las subunidades β o α, no se presentan fenotipos clínicamente diferentes.
La mutación Cis12Trp de la subunidad β1 (subunidad que se expresa en el cerebro, el esqueleto, el músculo y el corazón) interfiere con la capacidad de dicha subunidad para modular la cinética del canal de sodio; por ello decrece la tasa de inactivación de la subunidad alfa lo que causa incremento en el flujo de sodio; de esto resultan un mayor potencial de membrana e hiperexcitación (10); esta mutación cambia un residuo conservado de cisteína, dañando una unión disulfuro que normalmente mantiene un punto de unión extracelular a una inmunoglobulina (2,9). Al parecer la temperatura afecta la conductancia de los canales neuronales, que puede incrementarse en presencia de la mutación de la subunidad β1 (2).
Las mutaciones Thr875Met y Arg1648His de la subunidad α1 (subunidad que se expresa en las neuronas de los sistemas nerviosos central y periférico) del canal de sodio, se localizan en los segmentos 4 transmembrana de dicho canal (7,10), mientras que la mutación W1204R de la misma subunidad se encuentra en la segunda vuelta citoplasmática del segmento transmembrana 1 del dominio 3 (8). Estas mutaciones causan disminución en la tasa de inactivación del canal con incremento del flujo de Na+, produciendo aumento en la excitabilidad neuronal y en la susceptibilidad a las convulsiones (7). Estos cambios se dan en lugares del gen muy conservados en los vertebrados, o sea, poco susceptibles a mutaciones (8).
Se han descrito tres nuevas mutaciones para la subunidad α1: la mutación Tyr779Cys en el segmento 1 del dominio 2; las mutaciones Ile1944Thr y Met1841Thr localizadas en la región intracelular carboxilo terminal del canal de sodio; esta última se ha asociado con la Epilepsia Mioclónica Severa del Lactante (EMSL), de lo que podría inferirse que esta entidad hace parte de una presentación muy severa del espectro de la EGCF+, y no es una entidad distinta como se consideraba previamente (11,12).
Recientemente se han asociado dos nuevas mutaciones del SCN1A a la EGCF+ y a la Epilepsia Generalizada Tónico-Clónica Intratable de la Infancia (EGTCII) (Tabla N° 2). Este último síndrome epiléptico se caracteriza por crisis convulsivas recurrentes que se acomodan al fenotipo de la EGCF+ sin mioclonias o ausencias. Las crisis persisten con poca respuesta al tratamiento farmacológico, a diferencia de la EGCF+ en la que sí hay una respuesta adecuada; por este hecho la EGTCII constituye uno de los fenotipos más severos de la EGCF+ junto con la EMSL. Las dos mutaciones, T1709I ubicada entre los segmentos 5 y 6 del dominio 4, y V1611F en el segmento 3 del dominio 4 del canal de SCN1A, se han encontrado en dos individuos con EGTCII cuyas madres poseen un fenotipo clínico de EGCF+ y presentan la misma mutación en SCN1A. Otras mutaciones asociadas a la EGCF+ en SCN1A pueden verse en la tabla 2 (13).
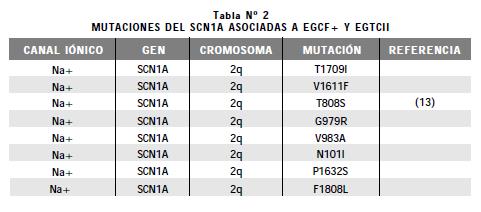
En la subunidad de SCN2A en 2q24 también sehan encontrado mutaciones asociadas a la EGCF+ (Tabla N° 1).
La mutación Arg187Trp de la subunidad α2 del canal de sodio dependiente del voltaje, se localiza en la vuelta citoplasmática entre los segmentos 2 y 3 del dominio 1; causa una inactivación más lenta que en el fenotipo nativo durante los potenciales de acción debido a una menor sensibilidad al voltaje, presumiblemente porque ha sido perturbado el acoplamiento entre el sensor de inactivación y el potencial transmembrana. Lo anterior produce efectos similares a los de las mutaciones para β1y α1 (6).
Mutaciones del canal de cloro unido a GABA
LA SUBUNIDAD γ2 se encuentra codificada en el gen GABRG2 del locus 5q y tres de sus mutaciones se han asociado a EGCF+: el cambio K289M entre el segundo y el tercer dominios transmembrana, relacionado con disminución en la amplitud de las corrientes del canal de cloro inducidas por GABA (14); la mutación R43Q que sustituye un residuo aminoácido altamente conservado en el primero de dos dominios de unión a benzodiacepinas (15); por último, el cambio en Q351 entre el tercero y cuarto dominios transmembrana, que induce un codón de parada en la proteína madura, con pérdida de la respuesta al GABA y, por lo tanto, incremento de la excitabilidad neuronal y las convulsiones (compatibles con la típica EGCF+); la consecuencia funcional de esta última mutación también se vio relacionada con la presencia de los tipos nativos de las subunidades α y β, es decir, esta subunidad γ2 mutada, interfiere con el complejo funcional del canal de cloro (16).
En conclusión, la EGCF+ es un síndrome cuya etiología se ha asociado a mutaciones de los canales de sodio dependientes del voltaje y de los canales neuronales de cloro; el cuadro clínico se presenta desde los 3 meses de edad y persiste más allá de los 6 años. Por su gran variabilidad fenotípica, otras epilepsias clasificadas de manera independiente pueden ser incluidas dentro de este síndrome, siempre y cuando su tiempo de aparición corresponda al de la EGCF+; el descubrimiento de mutaciones comunes para la EGCF+, la EMSL y la EGTCII hace pensar que el espectro de la primera es más amplio de lo que se ha pensado hasta la fecha, razón por la cual sería de gran utilidad el estudio de la EGCF+ en relación a otros síndromes epilépticos con fenotipos similares. Nuevos estudios podrían llevar a una reclasificación de esta entidad, además de entender su magnitud, su etiología y su fisiopatología, así como a obtener terapias farmacológicas más efectivas para el manejo de las crisis epilépticas.
BIBLIOGRAFÍA
1. CAMPOS J, CANELÓN M, GARCÍA M. Aspectos clínicos de las canalopatías epilépticas. Rev Neurol, 2000; 30 (Supl 1): S 42-S46. [ Links ]
2. WALLACE RH, WANG DW, SINGH R, SCHEFFER IE, GEORGE JR AL, PHILLIPS HA, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta 1 subunit gene SCN1B. Nat Genet, 1998; 19: 366-370. [ Links ]
3. KÖHLING R. Invited review: Voltage-gated sodium channels in epilepsy. Epilepsia, 2002; 43: 1.278-1.295. [ Links ]
4. SCHEFFER IE, BERKOVIC SF. Generalizad epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain, 1997; 120: 479-490. [ Links ]
5. BAULAC S, GOURFINKEL I, PICARD F, ROSENBERG M, PRUD'HOMME JF, BAULAC M, et al. A second locus for familial generalizad epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet, 1999; 65: 1.078-1.085. [ Links ]
6. SUGAWARA T, TSURUBUCHI Y, LAL AGARWALA K, ITO M, FUKUMA G, MAZAKI E, et al. A missense mutation of the Na+ channel alfa II subunit gene Nav 1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. PNAS, 2001; 98: 6.384-6.389. [ Links ]
7. ESCAYG A, MACDONALD BT, MEISLER MH. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet, 2000; 24: 343-345. [ Links ]
8. ESCAYG A, HEILS A, MACDONALDS BT, HAUG K, SANDER T, MEISLER MH. A novel SCN1A mutation associated with Generalizad Epilepsy with Febrile Seizures Plus – and prevalence of variants in patients with epilepsy. Am J Hum Genet, 2001; 68: 866-873. [ Links ]
9. WALLACE RH, SCHEFFER IE, PARASIVAM G, BARNETT S, WALLACE GB, SUTHERLAND GR, et al. Generalized epilepsy with febrile seizures plus: Mutation of the sodium channel subunit SCN1B. Neurology, 2002; 58: 1.426- 1.429. [ Links ]
10. WALLACE RH, SCHEFFER IE, RICHARDS M, DIBBENS L, DESAI RR, LERMAN T, et al. Neuronal sodium-channel alfa1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet, 2001; 68: 859-865. [ Links ]
11. ANNESI G, GAMBARDELLA A, CARRIDEO S, INCORPORA G, LABATE A, PASQUA AA, et al. Two novel SCN1A missense mutations in Generalizad Epilepsy with Febrile Seizures Plus. Epilepsia, 2003; 44: 1.257-1.258. [ Links ]
12. GAMBARDELLA A, CARRIDEO S, INCORPORA G, LABATE A, CIVITELLI D, POLIZZI A, et al. Three novel SCN1A missense mutations in Generalizad Epilepsy with Febrile Seizures Plus. Epilepsia, 2003; 44 (Suppl) 8: 50. [ Links ]
13. FUJIWARA T, SUGAWARA T, MAZAKI E, TAKAHASHI Y, FUKUSHIMA K, WATANABE M, et al. Mutation of sodium channel á subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain, 2003; 126: 531-546. [ Links ]
14. BAULAC S, HUBERFELD G, GOURFINKEL I, MITROPOULOU G, BERANGER A, PRUD'HOMME JF, et al. First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the gamma 2- subunit gene. Nat Genet, 2001; 28: 46-48. [ Links ]
15. WALLACE RH, MARINI C, PETROU S, HARBIN L, BOWSER DN, PANCHAL RG, et al. Mutant GABAA receptor gamma 2- subunit in childhood absence epilepsy and febrile seizures. Nat Genet, 2001; 28: 49-52. [ Links ]
16. HARKIN LA, BOWSER DN, DIBBENS LM, SINGH R, PHILLIPS F, WALLACE RH, et al. Truncation of the GABAA-receptor gamma 2 subunit in a family with Generalizad Epilepsy with Febrile Seizures Plus. Am J Hum Genet, 2002; 70: 530-536. [ Links ]
Recibido: 12 de febrero de 2004
Aceptado: 16 de abril de 2004

