










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.18 no.1 Medellín Jan./Mar. 2005
PRESENTACIÓN DE CASOS
Displasia diastrófica: Caracterización clínica, radiológica, citogenética y molecular de una paciente
Diastrophic dysplasia: clinical, radiological, cytogenetic and molecular characterization of a patient
José Luis Ramírez Castro1; Alicia María Cock Rada1; Beatriz Eugenia Mora Henao1; Gloria C. Ramírez Gaviria1; Gonzalo Vásquez Palacio1; Ana Elvira Prada Quiroz2
1 Unidad de Genética Médica, Facultad de Medicina, Universidad de Antioquia
2 Departamento de Radiología, Hospital Universitario San Vicente de Paúl, Medellín
RESUMEN
La displasia diastrófica (DTD) es un tipo de osteocondrodisplasia congénita con un patrón de herencia autosómico recesivo, descrita inicialmente por Lamy y Maroteaux en 1960. Se caracteriza por marcada disminución de la talla, normocefalia, micromelia, cifoescoliosis, paladar hendido, quistes del pabellón auricular, pie equino varo, contracturas articulares y pulgares abducidos. Durante el período neonatal la mortalidad es alta (25%), generalmente causada por obstrucción de la vía aérea. Si los pacientes sobreviven a dicho período, el pronóstico es bueno. El desarrollo mental y sexual es normal. En 1991 Hastbacka et al. identificaron el locus de la DTD en la región 5q31–q34. En 1996 el Treacher Collins Syndrome Collaborative Group ubicó el gen DTDST en la región 5q32–q33.1. Este gen codifica una proteína transportadora de sulfato, cuya alteración conduce a la formación de proteoglicanos poco sulfatados en la matriz cartilaginosa.
Presentamos una paciente que reúne las características fenotípicas y radiológicas de esta entidad. En el estudio molecular (Hospital Necker, París) se identificaron dos mutaciones en el gen DTDST; una mutación nueva y la otra ya descrita en la literatura.
PALABRAS CLAVE: enanismo diastrófico, gen dtdst, gen slc26a2, osteocondrodisplasia
SUMMARY
Diastrophic dysplasia is an osteochondrodysplasia that is inherited in an autosomal recessive pattern. It was first described by Lamy and Maroteaux in 1960. This disorder is characterized by short stature, normal head size, micromelia, kyphoscoliosis, cleft palate, cystic masses in auricles, talipes equinovarus, joint contractures and abduction of thumbs ('hitchhiker thumbs'). During the neonatal period mortality is high (25%), generally due to respiratory obstruction. For those who survive, prognosis is usually good. Intelligence and sexual development are normal. In 1991 Hästbacka et al. identified the DTD locus in the 5q31–q34 region. In 1996 the Treacher Collins Syndrome Collaborative Group showed that the gene is located on 5q32–q33.1. The DTDST gene encodes a sulphate transporter, whose alteration results in the production of undersulphated proteoglycans in the cartilage matrix.
We present a female patient who shows the phenotypic and radiologic characteristics of this disorder. Molecular studies (Necker Hospital, Paris) identified two mutations in the DTDST gene: one previously reported in the literature and a novel mutation.
KEY WORDS: diastrophic, dtdst gene, osteochondrodysplasia, slc 26 a2 gene
INTRODUCCIÓN
La displasia diastrófica (DTD) es un tipo de osteocondrodisplasia congénita con un patrón de herencia autosómico recesivo.1 En general las osteocondrodisplasias son alteraciones del sistema esquelético que producen trastornos de la remodelación ósea y del cartílago y/o del crecimiento.2
La DTD fue descrita inicialmente en 1960 por Lamy y Maroteaux. Antiguamente, en forma errónea, se les asignaba a estos pacientes el diagnóstico de artrogriposis múltiple congénita o de acondroplasia asociada con pie equino varo. Lamy y Maroteaux utilizaron el término 'diastrófico' proveniente del griego, que significa distorsionado, para referirse a la alteración esquelética propia de esta entidad.3
Las manifestaciones clínicas de la DTD pueden ser muy variables de un individuo afectado a otro y en ocasiones son tan leves que hacen muy difícil el diagnóstico, especialmente en neonatos que constituyan el primer caso en una familia. 4
Los hallazgos clínicos más frecuentes en esta entidad son: marcada disminución de la talla (dos o más desviaciones estándar por debajo del promedio poblacional), normocefalia, paladar hendido, quistes del pabellón auricular con calcificaciones del cartílago (oreja en coliflor) y micromelia (principalmente rizomelia y mesomelia). Además se observan escoliosis, cifosis cervical, hipoplasia de los cuerpos vertebrales cervicales, lordosis lumbar, calcificaciones prematuras de los cartílagos costales, espina bífida oculta, pie equino varo bilateral, contracturas articulares con cambios degenerativos progresivos, braquidactilia, pulgares abducidos e hipermóviles y sinfalangismo. Los cartílagos de la laringe, tráquea y bronquios son anormalmente blandos, lo cual puede contribuir al estrechamiento de las vías aéreas, al llanto de tono grave y a la voz aguda característicos de estos pacientes.5
Durante el período neonatal la mortalidad puede alcanzar hasta el 25% debido principalmente a obstrucción de las vías aéreas y neumonía por aspiración. También se han informado pacientes con cardiopatías congénitas asociadas que aumentan la mortalidad perinatal. Para quienes sobreviven a este período, el pronóstico de vida es bueno y casi siempre presentan inteligencia y desarrollo sexual normales. Sin embargo, en algunos pacientes pueden ocurrir complicaciones neurológicas debido a las anomalías en la columna cervical. Las alteraciones articulares generalmente son refractarias al tratamiento ortopédico. 5
Se ha logrado hacer diagnóstico prenatal mediante la ultrasonografía durante el segundo trimestre del embarazo, basándose en el acortamiento de las extremidades, la abducción de ambos pulgares y el pie equino varo. 6–8
Las características radiológicas de la DTD son: acortamiento y engrosamiento de los huesos tubulares con ensanchamiento de las metáfisis y aplanamiento irregular de las epífisis. Estas pueden presentar mineralización tardía. Los huesos del carpo generalmente presentan osificación acelerada (borde denso) en comparación con los demás huesos de la mano. Los metacarpos son cortos, gruesos e irregulares y pueden mostrar asimetría debido a la falta de desarrollo de alguna falange. La abducción bilateral del pulgar y del hallux y el pie equino varo son muy característicos. Se pueden observar coxa vara y genu valgo con dislocación lateral de la patela. La columna puede presentar escoliosis, espina bífida oculta desde C3–C4 hasta las vértebras torácicas superiores, hipoplasia de los cuerpos vertebrales cervicales, con cifosis y subluxación, estrechamiento de los espacios interpediculares de L1 a L5 y rotación posterosuperior del sacro. 9–11
La DTD es una entidad infrecuente; no obstante en Finlandia el 1–2% de la población es portadora de una mutación en el gen de la DTD (DTDST). 12
CASO CLÍNICO
Paciente evaluada al mes de edad por pie equino varo bilateral, contractura de los dedos de ambas manos, colección de líquido en las orejas y llanto de tono grave.
Hija de padres no consanguíneos. Producto del primer embarazo, sin complicaciones gestacionales. Nació de 38 semanas. Parto normal, en vértice, espontáneo. Al nacer: peso: 2.650 g, talla: 42 cm. Sin antecedentes familiares de casos similares.
Examen físico: talla 50 cm, PC 37 cm. Cráneo normocéfalo, fontanela anterior permeable, región frontal prominente, colecciones de líquido en los pabellones auriculares en proceso de reducción y las demás estructuras de la cara normales. Cuello corto. Tórax: mamilas invertidas. Cardiopulmonar, abdomen y genitales externos normales. Extremidades superiores: acortamiento rizomesomélico, desviación cubital de ambas manos, pulgares abducidos con ausencia del pliegue proximal de flexión y clinodactilia del quinto dedo en ambas manos. Extremidades inferiores: braquidactilia y pie equino varo bilateral (Figuras Nº 1 y 2).
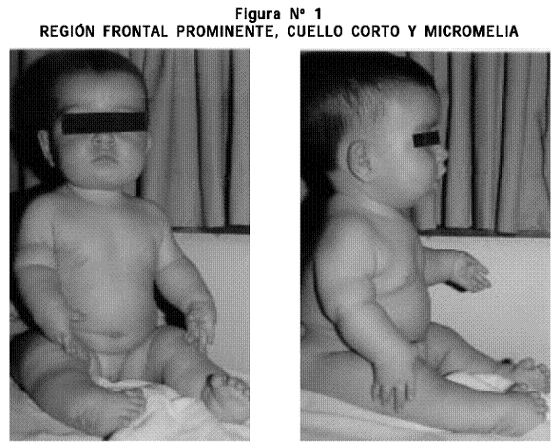
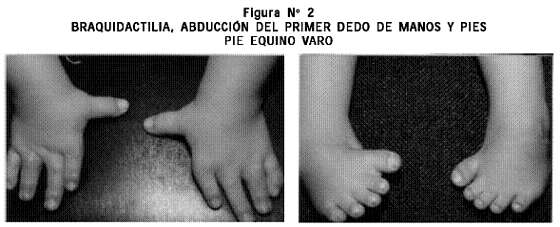
EXAMENES PARACLÍNICOS
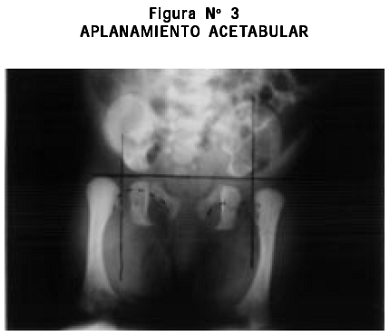
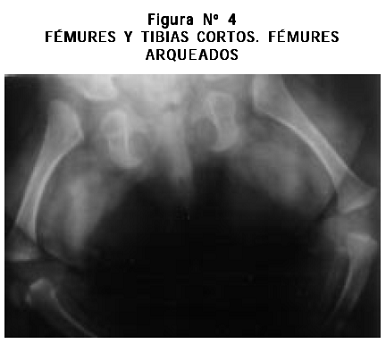
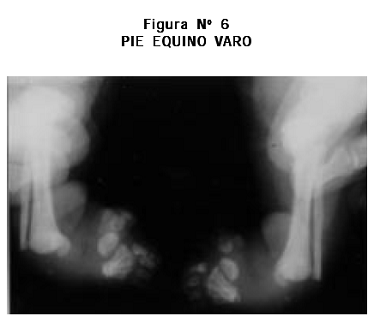
Radiografías: cifoescoliosis; cúbitos, radios, fémures y tibias cortos, leve arqueamiento de radios y tibias, aplanamiento acetabular, metacarpianos y metatarsianos cortos y engrosados bilateralmente (Figuras Nº 3, 4, 5, 6).

Cariotipo: 46, XX / 46 XX, fra (10q) (Figura Nº 7)

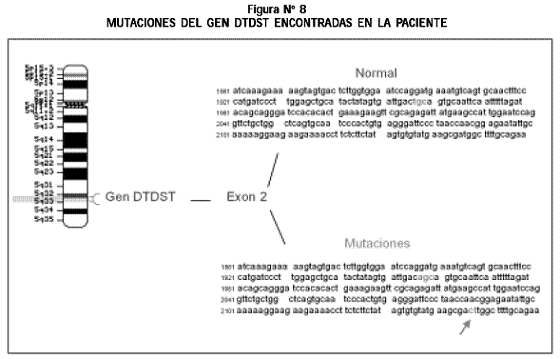
Estudio molecular: se detectaron mutaciones del gen DTDST (Diastrophic Dysplasia Sulfhate Transporter): una inserción de CT en el exón 2, codón 707, base 2147, heredada de la madre y una mutación en el exón 2, codón 653 (TGC por AGC) (C653S), heredada del padre (Figura Nº 8).
DISCUSIÓN
Las mutaciones en el gen DTDST son responsables de cuatro condrodisplasias que tienen un patrón de herencia autosómico recesivo; displasia diastrófica (DTD), displasia epifisiaria múltiple (DEM), atelosteogénesis tipo 2 (AO–II) y acondrogénesis 1B (ACG–1B). La ACG–1B es la más grave de estas condrodisplasias y por lo general es letal antes del nacimiento o inmediatamente después. En la AO–II también ocurre con frecuencia la muerte en el período neonatal, mientras que la DTD y la DEM se consideran las formas menos graves. 13–16
En 1991 Hästbacka et al. identificaron el locus de la DTD en la región 5q31–q34. 17 En 1996 el Treacher Collins Syndrome Collaborative Group indicó que el locus para el gen DTDST se ubicaba en la región 5q32–q33.1. Hasta el momento no hay evidencia de heterogeneidad de locus. 3
Durante la formación del sistema esquelético gran parte del hueso se origina a partir de cartílago (osificación endocondral). En adultos el cartílago está presente en las costillas, en el pabellón auricular, en las vías aéreas y en las articulaciones, donde tiene una función de soporte. El cartílago es esencial para el crecimiento y el mantenimiento del sistema esquelético humano. Sus características biológicas dependen principalmente de la matriz cartilaginosa, que está compuesta por colágeno específico del cartílago y por proteoglicanos sulfatados. 18 El gen DTDST, también denominado SLC26A2, codifica una proteína aniónica de membrana, transportadora de sulfato, que es necesaria para la sulfatación de los proteoglicanos que conforman la matriz cartilaginosa. El sulfato es importante para el cartílago porque aumenta su carga negativa, lo cual contribuye a sus propiedades de absorción de fuerzas. 19
El único tejido que se observa afectado por mutaciones en el gen mencionado es el cartílago; sin embargo, se ha encontrado su expresión en otros tejidos. Hailaa et al. 19 utilizaron técnicas de inmunohistoquímica e hibridación in situ para determinar la expresión de este gen y observaron una fuerte señal en el cartílago hialino fetal hipertrófico y en los condrocitos en proliferación, mientras que en los condrocitos primitivos de reserva la señal fue negativa. Además, se encontró expresión en las glándulas bronquiales, en el epitelio traqueal, en el tercio superior de las criptas colónicas, en las glándulas sudoríparas ecrinas, en el páncreas exocrino y en las vellosidades placentarias. Los testículos, el timo y la próstata no mostraron expresión. Una posible explicación para que las manifestaciones clínicas de la mutación del SLC26A2 se limiten al cartílago y al hueso podría ser que la función residual de la proteína SLC26A2 mutada, combinada con fuentes alternas de sulfato, sean suficientes para suplir las necesidades menores de otros tejidos diferentes al cartílago. 19
En la literatura se han informado más de 30 mutaciones y se ha observado correlación entre el genotipo y el fenotipo. Las mutaciones que producen una proteína truncada o una sustitución no conservativa de un aminoácido en el dominio trasmembranal, llevan a fenotipos graves, mientras que las sustituciones de un aminoácido no trasmembranal o en el sitio de corte producen fenotipos más leves. 20
El fenotipo clínico depende en parte del grado de actividad residual de la proteína transportadora de sulfato. Los individuos con ACG–1B, la forma más grave de las cuatro condrodisplasias, tienden a ser homocigotos para las mutaciones nulas (tasas de transporte de sulfato < 10% del cRNA salvaje) y los que presentan AO–II generalmente tienen al menos uno de los alelos con una mutación nula. Los individuos que manifiestan las formas mas leves, DTD y DEM, tienden a ser homocigotos para alelos con función residual, ya sea intermedia (tasas de transporte entre 10 y 50%) o casi normal (entre 50 y 100%). Sin embargo, no siempre se establece esta correlación entre la gravedad de la enfermedad y la función de la proteína, lo que sugiere que factores ambientales o la incapacidad del condrocito para utilizar vías alternas de transporte de sulfato contribuyen también a la variación en las características clínicas, lo cual es compatible con la expresión variable observada entre hermanos. 13
Como la DTD es especialmente prevalente en Finlandia, en 1991 Hästbacka et al. 12 identificaron una mutación en esa población que denominaron la mutación finlandesa fundadora, DTDST(Fin), puesto que se detectó en casi el 90% de los finlandeses con DTD. Esta mutación consiste en una transición de GTá GC en el sitio de corte donante de un exón 5' no traducido del gen DTDST que reduce de manera grave los niveles de mRNA. 12
Nuestra paciente muestra claramente las características fenotípicas y radiológicas de esta entidad. El estudio citogenético reveló una fragilidad del brazo largo del cromosoma 10, la cual no ha sido previamente informada en la literatura. El estudio molecular, realizado en el Hospital Necker de París, muestra dos mutaciones en el gen DTDST. La mutación heredada del padre, C653S, tiene una actividad casi igual a la del tipo salvaje, según estudios realizados por Karniski. 13 La mutación heredada de la madre aún no ha sido descrita en la literatura.
REFERENCIAS BIBLIOGRÁFICAS
1. HÄSTBACKA J, DE LA CHAPELLE A, MAHTANI MM, CLINES G, REEVE–DALY MP, DALY M, et al. The Diastrophic Dysplasia gene encodes a novel sulfate transporter: positional cloning by fine structure linkage disequilibrium mapping. Cell 1994; 78: 1.073–1.087. [ Links ]
2. HÄSTBACKA J, KAITILA I, SISTONEN P, DE LA CHAPELLE A. Diastrophic Dysplasia gene maps to the distal long arm of chromosome 5. Proc Natl Acad Sci USA 1990; 87: 8.056–8.059. [ Links ]
3. MCKUSICK VA. Online mendelian inheritance in man. Number 222600. Baltimore: The Johns Hopkins University; 1966–2004. [ Links ]
4. HALL BD. Diastrophic Dysplasia: extreme variability within a sibship. Am J Med Genet 1996; 63: 28–33. [ Links ]
5. JONES KL. Smith's Recognizable patterns of human malformation, 5th ed. Philadelphia: WB Saunders; 1997. [ Links ]
6. GOLLOP TR, EIGIER A. Prenatal ultrasound diagnosis of Diastrophic Dysplasia at 16 weeks. Am J Med Genet 1987; 27: 321–324. [ Links ]
7. SEVERI FM, BOCCHI C, SANSEVERINO F, PETRAGLIA F. Prenatal ultrasonographic diagnosis of Diastrophic Dysplasia at 13 weeks of gestation. J Matern Fetal Neonatal Med 2003; 13: 282–284. [ Links ]
8. WAX JR, CARPENTER M, SMITH W, GRIMES C, PINETTE MG, BLACKSTONE J, et al. Second–trimester sonographic diagnosis of Diastrophic Dysplasia: report of 2 index cases. J Ultrasound Med 2003; 22: 805–808. [ Links ]
9. TAYBI H. Diastrophic Dwarfism. Radiology 1963; 80: 1–10. [ Links ]
10. CAFFEY J. Pediatric X–Ray diagnosis, 7th ed. Chicago: Year Book Medical Publishers; 1978. [ Links ]
11. REMES VM, MARTTINEN EJ, POUSSA MS, HELENIUS IJ, PELTONEN JI. Cervical spine in patients with Diastrophic Dysplasia – radiographic findings in 122 patients. Pediatr Radiol 2002; 32: 621–628. [ Links ]
12. HÄSTBACKA J, KERREBROCK A, MOKKALA K, CLINES G, LOVETT M, KAITILA I, et al. Identification of the Finnish founder mutation for Diastrophic Dysplasia (DTD). Eur J Hum Genet 1999; 7: 664–670. [ Links ]
13. KARNISKI LP. Mutations in the Diastrophic Dysplasia sulfate transporter (DTDST) gene: correlation between sulfate transport activity and chondrodysplasia phenotype. Hum Mol Gen 2001; 10: 1.485–1.490. [ Links ]
14. CORSI A, RIMINUCCI M, FISHER LW, BIANCO P. Achondrogenesis type IB: agenesis of cartilage interterritorial matrix as the link between gene defect and pathological skeletal phenotype. Arch Pathol Lab Med 2001; 125: 1.375–1.378. [ Links ]
15. NEWBURY–ECOB R. Atelosteogenesis type 2. J Med Genet 1998; 35: 49–53. [ Links ]
16. HUBER C, ODENT S, RUMEUR S, PADOVANI P, PENET C, CORMIER–DAIRE V, et al. Sulphate transporter gene mutations in apparently isolated club foot. J Med Genet 2001; 38: 191–193. [ Links ]
17. HÄSTBACKA J, SISTONEN P, KAITILA I, WEIFFENBACH B, KIDD KK, DE LA CHAPELLE A. A linkage map spanning the locus for Diastrophic Dysplasia (DTD). Genomics 1991; 11: 968–973. [ Links ]
18. SATOH H, SUSAKI M, SHUKUNAMI C, IYAMA K, NEGORO T, HIRAKI Y. Functional analysis of Diastrophic Dysplasia sulfate transporter. J Biol Chem 1998; 273: 12.307–12.315. [ Links ]
19. HAILAA S, HÄSTBACKA J, BÖHLINGB T, KARJALAINENLINDSBERGB ML, KEREA J, SAARIALHO–KERED U. SLC26A2 (Diastrophic Dysplasia sulfate transporter) is expressed in developing and mature cartilage but also in other tissues and cell types. J Histochem Cytochem 2001; 49: 973–982. [ Links ]
20. ROSSI A, SUPERTI–FURGA A. Mutations in the Diastrophic Dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat 2001; 17: 159–171. [ Links ]
Recibido: 01 de septiembre de 2004
Aceptado: 03 de octubre de 2004

