










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.18 no.3 Medellín July/Sept. 2005
ARTÍCULO DE INVESTIGACIÓN
Bioequivalencia de dos formulaciones de metformina, tabletas de 850 mg, en voluntarios sanos colombianos*
Bioequivalence of two metformin formulations: 850 mg tablets in healthy colombian volunteers
Fanny Cuesta González1; Gloria Holguín Martínez2; Rosendo Archbold Joseph3; Adriana Ruiz Correa4; Margarita María Restrepo Garay5; Lina Peña Acevedo6; Blanca Montoya Beltrán7; Juan Carlos Ríos Toro8; Omar Correa Cano9
* Grupo de Estudio e Investigaciones Biofarmacéuticas
1. Ingeniera Química, Especialista en Farmacocinética.
2. Química Farmacéutica, Especialista en Sistemas de Calidad.
3. Químico Farmacéutico, Magíster en Educación.
4.Química Farmacéutica, Magíster en Ciencias Básicas Biomédicas, Doctora en Ciencias Farmacéuticas.
5. Química Farmacéutica, Magíster en Ciencias Básicas Biomédicas.
6. Médica Cirujana, Toxicóloga Clínica.
7.Bacterióloga y Laboratorista Clínica.
8.Médico y Cirujano, estudiante de Maestría en Ciencias Básicas Biomédicas.
9.Químico Farmacéutico. Especialista en Atención Farmacéutica, estudiante de Maestría en Ciencias Farmacéuticas.
RESUMEN
INTRODUCCIÓN: la metformina es un antihiperglicemiante útil en el manejo de la diabetes mellitus tipo II, del que se encuentran en el mercado colombiano tanto el producto innovador como diferentes formulaciones genéricas. Para garantizar la seguridad y eficacia de estas últimas, es necesario demostrar su bioequivalencia con respecto al producto innovador.
OBJETIVO: determinar si el producto Dimefor®/Metformina MK es bioequivalente con el producto Glucophage® (referencia) cuando se administran en dosis iguales a un grupo de voluntarios sanos.
MÉTODO: el estudio se realizó sobre veinticuatro voluntarios que cumplieron con los requisitos de inclusión y decidieron participar espontáneamente después de ser informados sobre su función en el estudio. Se utilizó un diseño aleatorio cruzado, en dos períodos, dos secuencias y doble ciego. Se administró una dosis única de 850 mg de cada producto y se tomaron muestras de sangre por un período de 24 horas. La cuantificación de la metformina se realizó por HPLC (High Performance Liquid Chromatography). Para la determinación estadística de bioequivalencia se utilizó la prueba de Schuirmann.
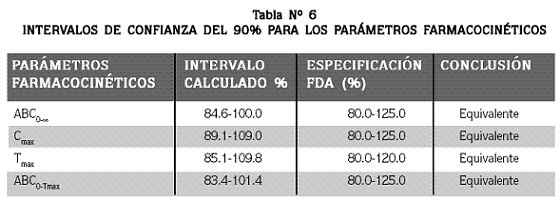
RESULTADOS: los dos productos fueron bioequivalentes con intervalos de confianza contenidos entre 80.0% y 125.0% para ln ABC0–∞ (84.6%–100.0%), ln Cmax (89.1%–109.0%) e ln ABC0–Tmax (83.4%–101.4%) y entre 80.0% y 120.0% para Tmax (85.1% y 109.8%).
PALABRAS CLAVE: biodisponibilidad, bioequivalencia, hplc (cromatografía líquida de alto desempeño), metformina
SUMMARY
INTRODUCTION: Metformin is an orally active antidiabetic agent used to treat type II diabetes; it is found in the Colombian market in both the innovator brand and the generic formulations. The latter have to prove some biopharmaceutical quality outcomes to guarantee interchangeable proprieties.
Objective: To determine whether the drug Dimefor®/Metformina MK is bioequivalent to the reference product Glucophage®, when the products are administrated, at the same dose, to a group of healthy volunteers.
METHOD: The study was made with 24 healthy volunteers who met the inclusion criteria and spontaneously decided to participate after being thoroughly informed. We used a two–sequence threeperiod randomized, crossed and double–blind study. The volunteers took an 850 mg dose of each medicine; then, blood samples were taken throughout 24 hours and the metformin quantification in plasma was determined by High Performance Liquid Chromatography with UV detection (HPLC/UV). For statistical analysis, Schuirmann's test was used.
RESULTS: The study showed that both preparations are bioequivalent; confidence intervals for ln AUC0–∞", ln Cmax, ln AUC0–Tmax and Tmax were [84.6–100.0%], [89.1–109.0%], [83.4–1.4%] and [85.1–109.8% ], respectively.
KEY WORDS: bioavailability, bioequivalency, hplc (high performance liquid chromatography), metformin
INTRODUCCIÓN
La bioequivalencia es la comparación de la velocidad y la magnitud a la cual un principio activo se absorbe a partir de una formulación de referencia (el producto innovador) y una formulación de prueba (producto genérico). Se evalúa mediante el análisis estadístico de parámetros farmacocinéticos obtenidos de los perfiles plasmáticos de los dos productos, administrados por vía oral, en voluntarios sanos. El parámetro farmacocinético que representa la magnitud de la absorción es el área bajo la curva (ABC), mientras que la velocidad es caracterizada por la concentración plasmática máxima (Cmax) y el tiempo en que se alcanza (Tmax). Cuando dos productos son bioequivalentes se espera que su eficacia y seguridad sean esencialmente las mismas, puesto que sus concentraciones plasmáticas a través del tiempo serán semejantes.
Las guías internacionales para estudios de bioequivalencia exigen que, antes de su evaluación, se establezca la equivalencia farmacéutica de las dos formulaciones. Para ello, ambos productos deben tener el mismo principio activo en cantidad semejante, de manera que al comparar la cantidad contenida en uno de ellos respecto al otro la diferencia no debe ser mayor de 5%; además, deben presentar igual forma de dosificación para ser utilizados por la misma vía de administración.1
La metformina es un fármaco antihiperglicemiante útil en el manejo de la diabetes mellitus tipo II, del que se pueden encontrar en el mercado colombiano tanto el producto innovador como diferentes formulaciones genéricas. Con el fin de garantizar la calidad biofarmacéutica de estas últimas, en cuanto a seguridad y eficacia, es necesario demostrar su bioequivalencia con respecto al producto innovador.
Desde el punto de vista farmacocinético, la absorción de la metformina administrada por vía oral es lenta e incompleta y ocurre principalmente en el intestino delgado. Se distribuye rápidamente a los tejidos corporales periféricos y prácticamente no se une a las proteínas plasmáticas. Tiene una biodisponibilidad del 50 al 60% y la Cmax se observa entre 2 y 4 horas después de la administración. No es metabolizada en el hígado o el tracto gastrointestinal, por lo que se excreta inalterada a través del riñón, con una vida media de eliminación que fluctúa entre 1.5 y 4.5 h.2
Teniendo en cuenta lo anterior, el objetivo de este estudio es determinar si el producto Dimefor®/Metformina MK es bioequivalente con el producto Glucophage® (referencia) cuando se administran en dosis iguales a un grupo de voluntarios sanos, siguiendo las recomendaciones dadas por el Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA),3 la Administración de Alimentos y Drogas de los Estados Unidos4 (FDA) y la Farmacopea Americana (USP),1 para este tipo de estudios.
MATERIALES Y MÉTODOS
Productos estudiados
Producto de prueba: Dimefor®/Metformina MK, tabletas de 850 mg elaborado por Tecnoquímicas S. A. (Cali, Colombia), lote 3P3510 con fecha de vencimiento octubre de 2005, suministrado por el laboratorio fabricante.
Producto de referencia: Glucophage®, tabletas de 850 mg elaborada por Merck Santè (Lyon, Francia), lote 104031 con fecha de vencimiento febrero del 2008, comprado por la Universidad de Antioquia en una farmacia.
Equivalencia farmacéutica
La valoración (contenido) del principio activo en las formulaciones y el ensayo de disolución se determinaron siguiendo los métodos suministrados por Tecnoquímicas S. A. Para la evaluación de la uniformidad de dosis se siguieron los lineamientos dados para este ensayo en la USP 28,1 utilizando el método de variación de peso. Durante las determinaciones analíticas, se utilizaron los siguientes materiales: estándar de metformina clorhidrato BP, lote 2 (European Pharmacopoeia, Estrasburgo, Francia), hidróxido de sodio y dihidrógeno fosfato de potasio, ambos grado reactivo, marca Merck (Darmstadt, Alemania), espectrofotómetro ultravioleta Shimadzu, modelo 1601 (Kioto, Japón), equipo de disolución Hanson Research, modelo SR2 (California, EE.UU).
Bioequivalencia
Voluntarios sanos
El estudio se realizó sobre veinticuatro sujetos, diecinueve hombres y cinco mujeres, seleccionados de la población estudiantil de la Universidad de Antioquia, con edades comprendidas entre 18 y 25 años (21.7 ± 1.6 años). El peso corporal de los voluntarios osciló entre 44.8–82.3 kg (61.8 ± 8.9 kg.) y la estatura entre 1.46 y 1.84 m (1.69 ± 0.09 m).
El proceso de selección se inició con aquellos sujetos, no fumadores, que decidieron participar espontáneamente después de ser informados sobre su función en el estudio. A continuación, se escogieron los que tenían una relación talla–peso dentro de ± 15% del peso ideal (Metropolitan Life Insurance Company, 1983).5 A este grupo se le practicaron exámenes físicos, médicos y de laboratorio. Entre los exámenes de laboratorio se evaluaron las funciones hematopoyética, hepática y renal, y se realizaron pruebas de VIH, hepatitis B y de embarazo en las mujeres. La muestra quedó conformada por voluntarios con resultados negativos en las pruebas mencionadas y que de acuerdo con la historia médica, los resultados del examen físico y de los exámenes de laboratorio estuvieran dentro de los rangos normales establecidos.
Dos semanas antes del inicio del estudio y durante el transcurso del mismo, los voluntarios dejaron de consumir medicamentos con o sin prescripción médica, incluidos los anticonceptivos orales. Una semana antes suspendieron el consumo de bebidas alcohólicas, al igual que el de bebidas o alimentos que contuvieran xantinas; esta restricción se mantuvo hasta la toma de la última muestra de sangre en cada período.
El día del estudio, los voluntarios permanecieron en ayuno 12 horas antes de la administración de la formulación. Una vez administrado el medicamento desayunaron y siguieron ingiriendo alimentos a los tiempos posadministración, en horas, que se describen a continuación: refrigerio, 2½; almuerzo, 5; refrigerio, 8, y cena, 11.
Preparación de los voluntarios y administración de las formulaciones
El día del tratamiento, a las 6:00 a. m, los voluntarios ingresaron a la Clínica de la Corporación para Estudios en Salud (CES). Inicialmente, a cada uno de ellos se le determinaron los signos vitales y se le recolectó una muestra de orina, para verificar la ausencia de alcohol y sustancias sicoactivas; luego, se procedió a insertarle un catéter en una vena del brazo y a tomar la primera muestra de sangre (t=0); inmediatamente después ingirió la dosis del medicamento correspondiente con 240 mL de agua.
Los sujetos permanecieron internados en la Clínica hasta la toma de la última muestra (24 h); durante este lapso de tiempo se registraron los efectos adversos observados por los responsables del estudio o informados por los voluntarios.
Diseño del estudio
La administración de las dos formulaciones a los voluntarios se llevó a cabo de acuerdo con un diseño aleatorio cruzado, en dos períodos, dos secuencias y doble ciego. En el período 1, doce sujetos recibieron la formulación de prueba y los doce restantes la de referencia; en el período 2, los voluntarios recibieron la formulación que no tomaron en el primero. Entre ambos períodos se dejaron transcurrir dos semanas.
El número de sujetos se calculó de acuerdo con la ecuación aplicada por Zapater6 y la FDA7 para este tipo de estudios, utilizando el promedio de los coeficientes de variación calculados de datos publicados en la literatura para este medicamento, 8–10 un poder estadístico del 80%, una magnitud mínima de diferencia que se debe detectar de 20%, un error β de 0.2 y un error α de 0.05.
Toma y tratamiento de las muestras
Las muestras (10 mL) se recolectaron en tubos con heparina (Becton Dickinson Vacutainer, New Jersey, EE.UU) a los siguientes tiempos, en horas: 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0, 6.0, 8.0, 10.0, 12.0, 14.0 y 24.0.
Una vez obtenidas las muestras, se centrifugaron (Indulap, Medellín, Colombia), a 2.000 rpm durante 15 minutos, se separó el plasma y se lo congeló a – 20ºC (Congelador Tecnifrío, Medellín, Colombia) hasta el día del análisis.
Técnica analítica
La concentración de metformina en las muestras se determinó teniendo en cuenta la técnica de extracción propuesta por Haley y col.11 y las condiciones cromatográficas usadas por Yuen y Peh,12 con algunas modificaciones. El principio activo se separó del plasma por extracción líquidolíquido con n–hexano, previa precipitación de las proteínas con acetonitrilo. La fase acuosa se secó, se redisolvió en fase móvil y se inyectó en un cromatógrafo líquido (Agilent, modelo HP 1100, California, EE.UU) con detector ultravioleta con arreglo de diodos (λ = 234 nm), controlado por el software ChemStation. La separación cromatográfica se realizó sobre una columna C18, de 4.0 x 250 mm, con partículas de 5 µm (Agilent, RPSelect B, California, EE.UU), utilizando como fase móvil una mezcla de acetonitrilo–buffer de dihidrógeno fosfato de potasio 0.01M, a pH 7.5 (60:40 v/v) a un flujo de 1.5 mL/min. Los reactivos utilizados fueron grado reactivo, exceptuando los solventes que se utilizaron en la fase móvil, que fueron calidad HPLC (JT Beaker, New Jersey, EEUU).
El método se validó antes del estudio de acuerdo con los lineamientos de la FDA.13 y los resultados mostraron que es específico, preciso, exacto y lineal en un rango de concentración entre 62.5 y 2.000 ng/mL de metformina.
Tratamiento de los datos
El análisis farmacocinético no compartimental de los datos se efectuó con el programa Microsoft® Excel 2000 (Microsoft Corporation, 1999) y los resultados se comprobaron con el programa PK solutions 2.0TM (Summit Research Services, 2004); para el análisis solo se tuvieron en cuenta aquellos valores superiores al límite de cuantificación del método analítico (62.5 ng/mL). Los parámetros farmacocinéticos que se calcularon fueron los siguientes: la concentración máxima (Cmax) y el tiempo necesario para alcanzarla (Tmax); la constante de eliminación (Ke), por regresión lineal de los pares de valores terminales del gráfico semilogarítmico de concentración plasmáticatiempo; el área bajo la curva de tiempo cero al último tiempo de muestreo (ABC0–t), por el método de los trapecios; el área bajo la curva de tiempo cero a infinito (ABC0–∞), mediante la suma de ABC0–t y el valor resultante de dividir la concentración plasmática encontrada en el último tiempo de muestreo por Ke; el área parcial bajo la curva de tiempo cero a la mediana de Tmax del producto de referencia (ABC0–Tmax), también por el método de los trapecios.
Análisis estadístico
Los datos transformados a logaritmos naturales, de ABC0–∞, ABC0–Tmax y Cmax y los de Tmax sin transformar, se compararon estadísticamente para determinar la bioequivalencia entre las dos formulaciones. Teniendo en cuenta el diseño utilizado, se realizó el análisis de los datos transformados (Statistica 6.0, Statsoft Inc. 2001) y se comprobaron los supuestos para el modelo del ANOVA en un diseño cruzado: efecto arrastre (carry–over) entre dos formulaciones; efecto de período y de formulaciones; variación intersujetos; variación intrasujetos, por medio de la prueba F ajustada de Pitman Morgan; normalidad de los residuos intersujetos e intrasujetos, aplicando la prueba de normalidad de Shapiro–Wilks; independencia de los residuos, y presencia de datos fuera de lo normal (outliers).
Para establecer la bioequivalencia se construyeron los intervalos de confianza del 90% con un nivel de significación de 0.05, utilizando el enfoque propuesto por Schuirmann (prueba de hipótesis unilateral doble).14 Las formulaciones se consideran bioequivalentes si los intervalos antes mencionados para el cociente de las medias de los datos transformados a logaritmos, de ABC0–∞ y ln Cmax, del producto de prueba con respecto al producto de referencia, están incluidos en el intervalo del 80.0% al 125.0%.
Ética
El protocolo del estudio fue aprobado por el Comité de Ética del Centro de Investigaciones Médicas de la Facultad de Medicina de la Universidad de Antioquia (Medellín, Colombia). Se realizó cumpliendo a cabalidad los principios de la Declaración de Helsinki y sus enmiendas,15 la Resolución Nº 008430/93 del Ministerio de Salud de Colombia16 y las Buenas Prácticas Clínicas (BPC).17 Todos los voluntarios firmaron el consentimiento informado antes del estudio y después de recibir información sobre su objetivo, las condiciones que debían cumplir, los posibles riesgos y los beneficios que obtendrían por su participación.
Resultados
Equivalencia farmacéutica
En la tabla Nº 1 se presentan los resultados de los ensayos de equivalencia farmacéutica, al igual que los límites (especificaciones) para cada uno de ellos.
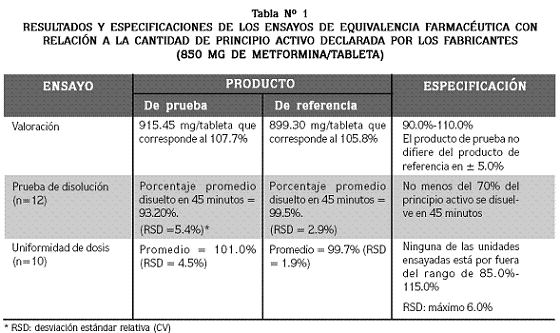
Aunque el ensayo de valoración del principio activo muestra que ambos productos contienen una cantidad ligeramente superior a la declarada por los fabricantes, esta se encuentra dentro de las especificaciones establecidas. La diferencia en el contenido de principio activo entre las dos formulaciones no es superior al 5% (1.9%). Los ensayos de disolución y uniformidad de dosis también se encuentran dentro de las especificaciones, lo que demuestra que los productos, además de ser equivalentes farmacéuticos, cumplen con las especificaciones de calidad farmacotécnica.
Bioequivalencia
Las concentraciones plasmáticas medias de metformina obtenidas para cada formulación, en cada tiempo de muestreo, se presentan en la tabla Nº 2.
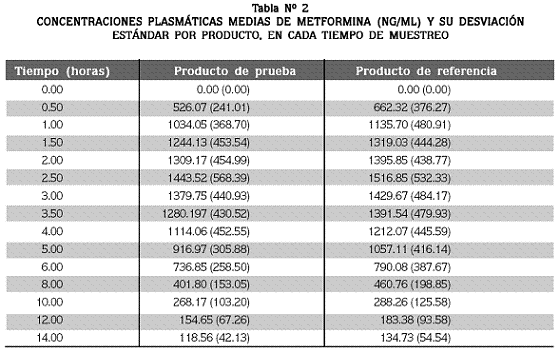
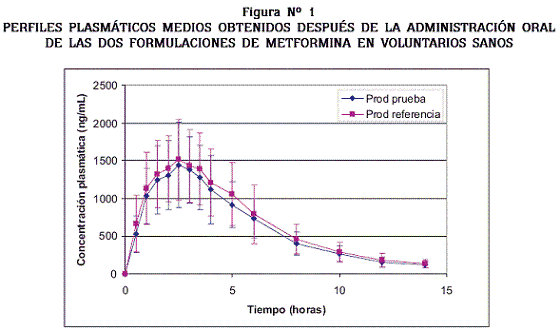
A pesar de que la toma de las muestras se realizó hasta las 24 horas, solo un voluntario, quien ingirió el producto de referencia, presentó un nivel plasmático (63.78 ng/mL) mayor que el límite de cuantificación del método, en este tiempo de muestreo. Si se comparan las concentraciones plasmáticas obtenidas para ambas formulaciones, se observa que la absorción es semejante, lo que se confirma con la similitud de los perfiles plasmáticos que se presentan en la Figura Nº 1.
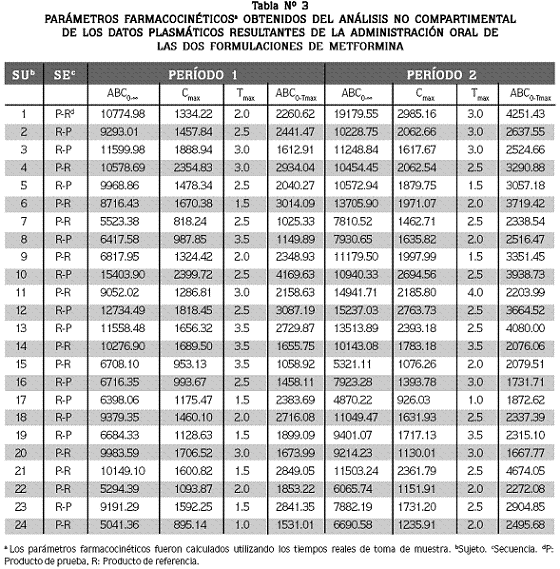
En la tabla Nº 3 se reúnen los parámetros farmacocinéticos obtenidos para cada voluntario, por análisis no compartimental de los datos plasmáticos. Se muestra adicionalmente la secuencia en la que los voluntarios ingirieron las formulaciones.
Los resultados del análisis de varianza de cada uno de estos parámetros se presentan en la tabla Nº 4.
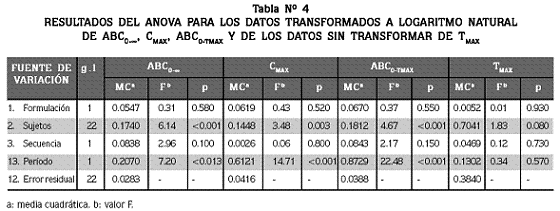
La evaluación de los supuestos del modelo probabilístico mostró que mientras no hubo efecto de arrastre entre las dos formulaciones para ninguno de los parámetros, sí se encontró efecto de período para todos ellos, excepto para Tmax. El efecto período se manifestó en valores medios mayores en el período 2 que en el período 1. De otro lado, para la variabilidad intersujetos se obtuvo una significancia alta (p<0.005), dando a entender que la población de la cual se eligieron los sujetos presenta una variabilidad muy elevada. Para evaluar la variación intrasujeto fue necesario hacer una corrección a la prueba ajustada de Pitman–Morgan, debido al efecto período encontrado. Los resultados mostraron que esta fue similar para las dos formulaciones en cada uno de los parámetros medidos.
El análisis de los residuos reveló que tanto los residuos intersujeto como los intrasujeto fueron normales e independientes con todos los parámetros. En cuanto a la presencia de datos fuera de lo normal, el voluntario uno presentó este tipo de valores para los parámetros ABC0–∞ y Cmax; estos fueron mucho más altos en el segundo período que en el primero (Tabla Nº 3).
En la tabla Nº 5 se reúnen las reacciones adversas que fueron informadas por los voluntarios o detectadas por el personal médico durante los dos períodos del estudio.
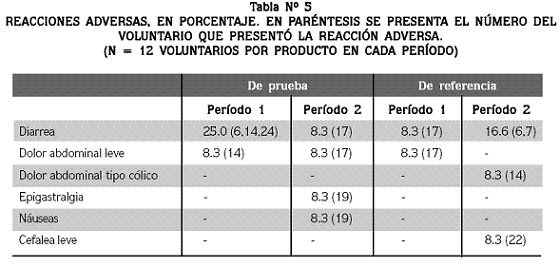
El efecto secundario más común fue la diarrea, la cual se presentó entre 2.5 y 6 h después de la administración del medicamento. Todos los voluntarios concluyeron el estudio.
DISCUSIÓN
Equivalencia farmacéutica
El estudio de equivalencia farmacéutica, además de garantizar que las formulaciones contienen el mismo principio en concentraciones semejantes, muestra que ambas cumplen los criterios básicos de calidad establecidos para formas sólidas de administración oral. En este sentido, el cumplimiento del ensayo de disolución garantiza que la formulación liberará el principio activo una vez ingerida, mientras que el ensayo de uniformidad de dosis demuestra que todas las tabletas del lote contienen la cantidad declarada de principio activo dentro de los límites establecidos. Sin embargo, el hecho de que los dos productos sean equivalentes farmacéuticos no garantiza que sean bioequivalentes.
Bioequivalencia
La absorción del principio activo fue rápida; a los 30 minutos se detectaron niveles plasmáticos importantes que permanecieron hasta 14 horas después de la administración. Las Cmax medias fueron de 1631.08 (± 535.70) ng/mL y 1643.42 (± 517.57) ng/mL para el producto de prueba y de referencia, respectivamente, en tanto que los tiempos para alcanzarlas fueron de 2.42 (± 0.75) h y 2.48 (± 0.70) h. Ambos parámetros fueron muy semejantes para las dos formulaciones. Los valores de Tmax, además, fueron similares a los obtenidos por otros autores.18,19
La extensión de la absorción fue similar: el ABC0–∞ medio para el producto de prueba fue de 9154.79 (± 2617.86) ng.h/mL y para el de referencia 10064.79 (± 3430.82) ng.h/mL y el área parcial media ABC0–Tmax para el producto de prueba fue de 2414.35 (± 809.18) ng.h/mL y para el producto de referencia de 2622.93 (± 911.68) ng.h/mL La similitud de las formulaciones también se refleja en los perfiles farmacocinéticos (Figura Nº 1) y se confirma con el análisis estadístico (Tabla Nº 6) que mostró que ambos productos no solo son bioequivalentes con respecto a los parámetros obligatorios (ABC0–∞ y Cmax), sino también con respecto a Tmax y ABC0–Tmax.
Este último, conocido como exposición temprana, es un parámetro propuesto por la FDA4 que proporciona una idea sobre el control de absorción del fármaco.
El análisis de los supuestos del ANOVA puso de manifiesto una gran variabilidad en la disposición de este fármaco por parte de los voluntarios que participaron en el estudio. Dicha variabilidad es evidente en las barras de error de los perfiles plasmáticos (Figura Nº 1) y también ha sido encontrada por otros autores.8,10 El número de sujetos necesario para llevar a cabo esta investigación fue calculado teniendo en cuenta esta consideración.
También se encontraron diferencias estadísticas entre los niveles plasmáticos obtenidos en el período 1 y el período 2. Aunque el diseño estadístico corrige esta eventualidad, se han revisado todos los procedimientos que se realizaron con el fin de encontrar la causa de esta situación; sin embargo, no ha sido posible establecerla.
En este estudio no se presentaron reacciones adversas serias; los eventos registrados se relacionaron principalmente con molestias gastrointestinales (Tabla Nº 5) que se resolvieron espontáneamente. Estos efectos colaterales son bastante comunes con este fármaco y han sido descritos por otros investigadores.18
CONCLUSIÓN
El análisis estadístico de los parámetros farmacocinéticos ABC0–∞, Cmax, Tmax y ABC0–Tmax, dio como resultado que las dos formulaciones sometidas al estudio son semejantes; por lo tanto, el producto de prueba, comercializado y producido por Tecnoquímicas S.A. como Dimefor® y Metformina MK, tabletas de 850 mg, con número de lote 3P3510, es bioequivalente al producto de referencia Glucophage®, tabletas de 850 mg de metformina, con número de lote 104031. Con estos resultados es posible inferir que los dos productos ejercerán un efecto terapéutico similar si se administran indistintamente a un paciente determinado, pudiendo, el prescriptor, tener elementos sólidos para decidir sobre la intercambiabilidad de estas formulaciones.
AGRADECIMIENTOS
Este estudio fue patrocinado por Tecnoquímicas S. A., contrato 8716–002 de 2004. Los autores agradecen al profesor Abel Díaz C., la asesoría estadística y al estudiante de Química Farmacéutica Iván Darío Gómez, la colaboración en la actividad clínica.
Los autores manifiestan no tener vínculos afectivos ni laborales con las empresas Lakor Farmacéutica S.A. y Merck.
REFERENCIAS BIBLIOGRÁFICAS
1. US Pharmacopeia – National Formulary. The official compendia of standards.(USP 26 / NF 21). Rockville: United States Pharmacopeial Convention Inc. 2003: 2.334–2.342. [ Links ]
2. DUNN CJ, PETERS DJ. Metformin. A review of its pharmacological properties and therapeutic use in non–insulin–dependent diabetes mellitus. Drugs 1995; 49: 721 –749. [ Links ]
3. Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA), Colombia. Guía de biodisponibilidad y bioequivalencia 1997. Acta 51. [ Links ]
4. Food and Drug Administration. Guidance for Industry. Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. 2003 [fecha de acceso 15 de mayo de 2003]. Disponible en: http://www.fda.gov/cder/guidance/index.htm. [ Links ]
5. Metropolitan Life Insurance Company. Ideal height and weight tables, 1983 [Fecha de acceso 15 de mayo de 2001]. Disponible en http://www.med.umich.edu/1libr/primry/life15.htm [ Links ]
6. ZAPATER P, HORGA JF. Bioequivalencia y genéricos. Los estudios de bioequivalencia I. Una aproximación a sus bases teóricas, diseño y realización. Rev Neurol 1999; 29: 1.235–1.246. [ Links ]
7. Food and Drug Administration. Guidance for Industry. Statistical Approaches to Establishing Bioequivalence. 2001 [fecha de acceso 20 de junio de 2001]. Disponible en: http://www.fda.gov/cder/guidance/index.htm. [ Links ]
8. CAILLE G, LACASSE Y, RAYMOND M, LANDRIAULT H, PRERROTA M, PICIRILLI G, et al. Bioavailability of metformin in tablet form using a new high pressure liquid chromatography assay method. Biopharm Drug Dispos 1993; 14: 257–263. [ Links ]
9. YUEN KH, WONG JW, BILLA N, JULIANTO T, TOHWT. Bioequivalence of generic metformin tablet preparation. Int J Clin Pharmacol Ther 1999; 37: 319–322. [ Links ]
10. NAJIB N, IDKAIDEK N, BESHTAWI M, BADER M, ADMOUR I, ALAM SM, et al. Bioequivalence evaluation of two brands of metformin 500 mg tablets (Dialon® and Glucophage®) in healthy human volunteers. Biopharm Drug Dispos 2002; 23: 301–306. [ Links ]
11. HALE TW, KRISTENSEN JH, HACKETT LP, KOHAN R, ILETT KF. Transfer of metformin into human milk. Diabetologia 2002; 45: 1.509–1.514. [ Links ]
12. YUEN KH, PEH KK. Simple high–performance liquid chromatographic method for the determination of metformin in human plasma. J Chromatogr B 1998; 710: 243–246. [ Links ]
13. Food and Drug Administration. Guidance for Industry. Bioanalytical Method Validation [fecha de acceso 20 de junio de 2001]. Disponible en http://www.fda.gov/cder/guidance/index.htm. [ Links ]
14. SCHUIRMANN DJ. A comparison of the two–one side test procedures and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm 1987; 15: 687–681. [ Links ]
15. World Medical Association (WMA). Declaration of Helsinki. Edinburgh: Ethical principles for medical research involving human subjects; October 2000. [ Links ]
16. Ministerio de Salud de Colombia. Resolución Nº 008430 de 1993. Bogotá: Ministerio de Salud de Colombia; 1993. [ Links ]
17. Organización Panamericana de la Salud – Organización Mundial de la Salud OPS/OMS. Normas Éticas Internacionales para las Investigaciones Biomédicas con Sujetos Humanos. Ginebra: OPS/OMS;1996. [ Links ]
18. CHEN CL, YU LX, LEE HL, YANG CY, LUE CS, CHOU CH. Biowaiver extension potential to BCS class III high solubility–low permeability drugs: bridging evidence for metformin immediate–release tablet. Eur J Pharm Sci 2004; 22: 297–304. [ Links ]
19. MARATHE PH, ARNOLD ME, MEEKER J, GREENE DS, BARBHAIYA RH. Pharmacokinetics and bioavailability of a metformin/glyburide tablet administered alone and with food. J Clin Pharmacol 2000; 40: 1494–1502. [ Links ]
Recibido: agosto 05 de 2005
Aceptado: agosto 23 de 2005

