










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.18 no.3 Medellín July/Sept. 2005
ARTÍCULO DE REVISIÓN
Importancia de la terapia génica en la enfermedad granulomatosa crónica
Gene therapy in chronic granulomatous disease
Luz Astrid Velásquez Marulanda1*; Julián Camilo Arango Rincón2*; Andrés Augusto Arias Sierra3*; Pablo Javier Patiño Grajales4*
* Grupo de Inmunodeficiencias Primarias, Corporación Biogénesis, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
1. Bacterióloga
2. Estudiante de Microbiología y Bioanálisis
3. Bacteriólogo, M.SC
4. MD, M.SC, D.SC*
Correspondencia: Pablo Javier Patiño. Sede de Investigación Universitaria (SIU). Calle 62 #52–59 Oficina 530 Universidad de Antioquia, Medellín, Antioquia, Colombia.
Teléfono: (574) 210 64 75, FAX: (574) 510 60 47
Correo electrónico: ppatino@udea.edu.co
RESUMEN
EL SISTEMA NADPH OXIDASA DE LAS CÉLULAS FAGOCÍTICAS es un complejo enzimático encargado de producir anión superóxido durante la respuesta contra los microorganismos. Mutaciones en los genes que codifican para las proteínas de este sistema son responsables de la Enfermedad Granulomatosa Crónica (EGC) que es una inmunodeficiencia primaria caracterizada por la presencia de infecciones recurrentes debidas a un grupo específico de microorganismos, principalmente oportunistas. Actualmente el tratamiento para la mayoría de los pacientes con EGC está dirigido a la prevención o al control de los procesos infecciosos, pero no a la curación de la enfermedad. El tratamiento curativo consiste en el trasplante alogénico de médula ósea (TMO); sin embargo, este método enfrenta dificultades como la incompatibilidad de HLA, la inmunosupresión debida a las condiciones mieloablativas necesarias para el trasplante y el riesgo de desarrollar la enfermedad injerto contra hospedero. Como una alternativa al TMO, ha surgido la terapia génica ex vivo en células progenitoras hematopoyéticas. Las características genéticas de la EGC le han permitido convertirse en un modelo para el estudio de la terapia génica ex vivo. En este artículo se describen y analizan los resultados que hasta la fecha se han obtenido en el campo de la terapia génica aplicada a la EGC.
PALABRAS CLAVE: enfermedad granulomatosa crónica, NADPH oxidasa, terapia génica, vectores retrovirales
SUMMARY
Reactive oxygen species (ROS) production by phagocytes is an important mechanism to kill invading microorganisms. Neutrophils from individuals with chronic granulomatous disease (CGD) do not produce ROS, thereby rendering these individuals more susceptible to infection. CGD results from mutations in the genes encoding essential subunits of respiratory burst NADPH oxidase, the enzyme complex necessary for the production of these reactive molecules. The absence of phagocyte ROS results in recurrent fungal and bacterial infections and inflammatory granulomas, associated with significant morbidity and mortality. Currently, the curative treatment is the allogenic bone marrow transplant (BMT); nevertheless, this therapy has some disadvantages including the HLA incompatibility, the immunosupression due to the myeloablative conditions necessary for the transplant and the high risk to develop graft vs. host disease. As an alternative to BMT the ex vivo gene therapy in hematopoietic stem cells has been intensely studied. Although this option could be the most appropriate treatment, it can give rise to other kinds of adverse effects. The genetic features of CGD have made it a very attractive candidate to be cured with gene therapy. This review summarizes and discusses the current advances about gene therapy and its application to CGD.
KEY WORDS: chonic granulomatous disease, gene therapy, nadph oxidase, retroviral vectors
EL SISTEMA NADPH OXIDASA
Las células especializadas en la fagocitosis tienen la capacidad de ingerir microorganismos en el interior de una vacuola fagocítica y destruirlos mediante varios mecanismos microbicidas, entre ellos los dependientes del oxígeno. Estos se relacionan con la producción de especies reactivas del oxígeno (ROS, por su sigla en inglés), derivadas del anión superóxido (O2–), generadas por la actividad catalítica del sistema dinucleótido de nicotinamida y adenina fosfato (NADPH) oxidasa.
Las ROS causan gran toxicidad a un amplio espectro de microorganismos lo cual les permite ejercer un papel sinérgico con el contenido de los gránulos para eliminar los microorganismos fagocitados. El sistema NADPH oxidasa está conformado por cinco proteínas; dos de estas, gp91phox y p22phox, se encuentran en la membrana celular o en la membrana de la vacuola fagocítica, formando un heterodímero llamado flavocitocromo b558, el cual posee dos grupos hemo y un grupo dinucleótido de flavina y adenina (FAD), necesarios para la transferencia de electrones durante la activación del sistema. Las tres proteínas restantes: p47phox, p67phox y p40phox, se encuentran localizadas en el citosol acompañadas de una GTPasa de bajo peso molecular conocida como Rac2. La actividad catalítica del sistema NADPH oxidasa, estimula la transferencia de electrones desde el NADPH disuelto en el medio citosólico, hacia el oxígeno molecular que se encuentra en el interior de la vacuola fagocítica dando lugar al anión superóxido (O2–). A partir de este radical se generan otros compuestos como el peróxido de hidrógeno (H2O2), el radical hidroxilo (OH–), el ácido hipocloroso (HClO) y halógenos, que participan en la destrucción de los microorganismos que fueron fagocitados (Figura Nº 1).1,2
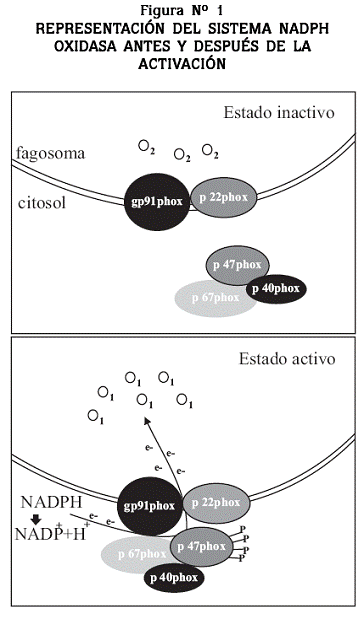
ENFERMEDAD GRANULOMATOSA CRÓNICA
La Enfermedad Granulomatosa Crónica (EGC) es una inmunodeficiencia primaria que resulta de la alteración en alguno de los componentes proteicos del sistema NADPH oxidasa (excepto p40phox), lo que ocasiona una marcada reducción o la inhibición total en la producción del anión superóxido.3–6 Los pacientes con EGC se caracterizan por la presencia de infecciones recurrentes de etiología bacteriana y fúngica, localizadas frecuentemente en la piel, el pulmón y el intestino. Sus manifestaciones clínicas comunes incluyen: linfadenitis, abscesos cutáneos y neumonía, así como diarrea, colitis y en ocasiones sepsis. Estas infecciones generalmente se complican con abscesos hepáticos, osteomielitis o con obstrucción en las vísceras huecas por el crecimiento de los granulomas tal como sucede en la cistitis granulomatosa y en los granulomas en el estómago y el esófago. Las bacterias y hongos involucrados comúnmente son la Salmonella sp, el Staphylococcus aureus, la Burkholderia cepacia y el Aspergillus fumigatus.6–8
La forma más frecuente de la EGC es la ligada al cromosoma X (EGC–X), originada por una mutación en el gen CYBB que codifica para el componente gp91phoxdel sistema NADPH oxidasa.9,10 La segunda forma más común es la debida a mutaciones en el gen NCF–1 que codifica para la proteína p47phox. 11 Luego se encuentra la alteración en el gen NCF–2, el cual codifica para la proteína p67phox.12 Por último, la forma menos común es la causada por mutaciones en el gen CYA, el cual codifica para la proteína p22phox.6,7
Diagnóstico de la EGC
El diagnóstico de la EGC se basa en la demostración de la incapacidad de respuesta de las células fagocíticas ante diferentes agentes activadores como el forbol miristato acetato (FMA).6,7,13,14
Tratamiento de la EGC
En la actualidad el tratamiento para los pacientes con diagnóstico de EGC se basa en el control de los procesos infecciosos y en la profilaxis antimicrobiana. La administración de interferón gama (IFNγ) está indicada como mantenimiento profiláctico y como terapia inmunomoduladora con la cual se ha demostrado una disminución en la recurrencia y gravedad de las infecciones.7,13,15
En el momento no existe un acuerdo con relación al trasplante de médula ósea alogénico como tratamiento curativo de la EGC, pues este procedimiento tiene limitaciones importantes tales como: la dificultad para encontrar un donante histocompatible, la alta probabilidad de desarrollar la enfermedad injerto contra hospedero (EIH) y la inmunosupresión generada por las condiciones mieloablativas previas al trasplante. De otro lado, la terapia génica ofrece la ventaja de ser un trasplante de carácter autólogo, lo que elimina las probabilidades de desarrollar EIH pero, aunque mejora el estado de inmunosupresión, puede dar origen a otras complicaciones como alergias, autoinmunidad y desarrollo de cáncer.6,7,16–21
LA TERAPIA GÉNICA EN CÉLULAS HEMATOPOYÉTICAS
La terapia génica es una técnica que busca introducir un gen funcional dentro de una célula determinada y de esta manera corregir un defecto genético o conferirle a la célula una función adicional o diferente a la que naturalmente tiene. En la terapia génica ex vivo se obtienen células del paciente para ser modificadas en el laboratorio y posteriormente implantarlas en el organismo con el defecto genético. Este método se emplea para realizar terapia génica en la EGC y en otros trastornos hematopoyéticos, como la inmunodeficiencia combinada severa (SCID por su sigla en inglés), el síndrome de Wiskott–Aldrich, la anemia de Fanconi y la anemia falciforme, entre otros.22,23
Las células diana para corregir el defecto genético en estas enfermedades son las células pluripotenciales hematopoyéticas. Estas células son escasas, representan el 0.01% de toda la población celular hematopoyética, y se caracterizan por encontrarse en la fase G0 del ciclo celular. Las células progenitoras hematopoyéticas humanas utilizadas en el campo de la terapia génica son seleccionadas por la expresión del antígeno CD34 y se puede inducir su proliferación mediante la administración de factores de crecimiento, de forma que se aumente considerablemente su número en la sangre periférica. La habilidad de estas células para repoblar la médula ósea de individuos receptores que han sido expuestos a condiciones mieloablativas, así como su capacidad para alcanzar la diferenciación en un linaje específico de poblaciones celulares en la sangre periférica, las convierte en un blanco importante para la intervención genética.22 Esto explica por qué las células madre ofrecen oportunidades terapéuticas importantes, a pesar de que es necesario profundizar en el conocimiento acerca de la biología y de los factores microambientales que influencian su desarrollo y proliferación in vivo e in vitro.24
Vectores utilizados en la terapia génica
Para introducir el gen terapéutico y así lograr el efecto deseado en las células diana, se han desarrollado varias estrategias; la de los vectores virales es la más utilizada actualmente. Los sistemas virales han demostrado tener mayor utilidad como vectores para estudios de terapia génica en la EGC, especialmente los construidos a partir de retrovirus como los oncorretrovirus y los lentivirus. Estos sistemas virales se han clasificado como virus ecotrópicos si infectan exclusivamente células murinas o virus anfotrópicos si infectan células de la mayoría de las especies incluyendo células humanas y murinas.25,26 Para que la infección por vectores oncorretrovirales, como es el caso de los derivados del virus de la leucemia murina (MLV, por su sigla en inglés) sea efectiva, es necesario que la célula esté en división, ya que la entrada del ADN viral al núcleo sólo puede ocurrir con la disolución de la membrana nuclear durante la mitosis. En contraste, la infección por vectores lentivirales, como los basados en el VIH (virus de la inmunodeficiencia humana), puede ocurrir en células que no se encuentran en división, gracias a señales de localización nuclear que permiten el transporte del genoma viral a través de la membrana nuclear.22
Para evitar que estos vectores virales tengan los efectos patógenos del virus original, su genoma ha sido modificado para convertirlos en herramientas de transferencia genética anulando su capacidad replicativa competente. Para lograr este propósito, se remueven las secuencias genómicas virales que les confieren características patógenas, disminuyendo así la probabilidad de replicación viral. El genoma de un retrovirus típico (figura Nº 2), contiene repeticiones terminales largas (LTR, por su sigla en inglés), secuencias para señales de encapsidación (Ψ) y secuencias como gag, pol y env, las cuales codifican para la transcriptasa reversa, la integrasa y proteínas de la envoltura viral.
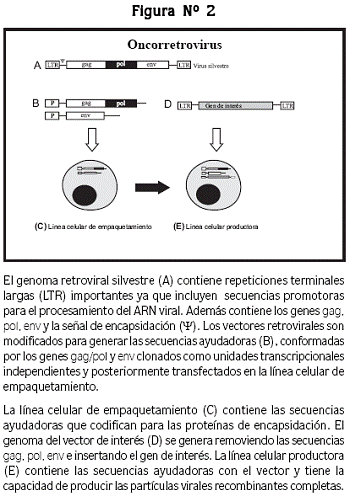
Cuando se remueven las secuencias gag, pol y env se genera un espacio que servirá para insertar el gen de interés, en forma de ADN complementario (cADN), el cual estará acompañado de las LTR y de la señal de encapsidación, pero al mismo tiempo el virus habrá perdido su capacidad replicativa. Las secuencias removidas se introducen en plásmidos como unidades transcripcionales independientes, para luego ser transfectadas en una línea celular que se denomina línea celular de empaquetamiento. Las células que contienen las proteínas necesarias para que se lleve a cabo la encapsidación del genoma viral, se transducen con el vector que contiene el gen terapéutico, lo que permitirá establecer una línea celular productora. Luego, entre estas secuencias ocurren reacciones de complementación (en trans) que permiten la formación de partículas virales recombinantes completas, que contienen en su interior el cADN del gen terapéutico.27,28 Otra modificación que se puede realizar en el vector consiste en la generación de virus quiméricos, es decir, virus que tienen la capacidad de empaquetar su genoma con las proteínas de envoltura de otros virus, con lo que aumentan su rango de células hospederas.29,30
Riesgos de los vectores retrovirales
Existen dos riesgos al utilizar vectores retrovirales: la posibilidad de producir mutagénesis insercional y la generación de virus silvestres. La mutagénesis insercional es un evento aleatorio que ocurre cuando el vector retroviral se inserta en un sitio de un gen celular en el que podría originar el desarrollo de procesos proliferativos anormales, por medio de la inactivación de genes supresores de tumor o por la activación de protooncogenes. Un ejemplo de esto se presentó en dos pacientes con inmunodeficiencia combinada grave, que luego de aproximadamente tres años de terapia sufrieron una expansión clonal desordenada de linfocitos T CD4 maduros.27,31–33 Con respecto a la generación de virus silvestres, este es un fenómeno que puede ser controlado con la eliminación de la homología entre los genes de empaquetamiento en el vector, así como con la separación de estos genes en dos o más unidades transcripcionales independientes, como ya se ha mencionado. Hasta el momento la ocurrencia de efectos adversos después de un tratamiento con terapia génica, solo puede controlarse con el seguimiento de la evolución clínica de los pacientes.34,35
La terapia génica en la EGC
Se ha llevado a cabo una gran variedad de estudios de transferencia génica en líneas celulares modelo de EGC, en ratones Knockout para proteínas del sistema NADPH oxidasa, en ratones modelo de SCID y en pacientes con EGC.18,19,36 Las células B transformadas con el virus de Epstein–Barr han sido utilizadas por muchos grupos de investigación como modelos in vitro para evaluar la corrección genética en EGC. Esta línea celular expresa cantidades pequeñas de los componentes proteicos del sistema NADPH oxidasa y puede producir bajos niveles de anión superóxido; sin embargo, hasta el momento se desconoce la importancia fisiológica de la generación del anión superóxido por parte de los linfocitos B.37,38 La capacidad de producir anión superóxido en estas líneas celulares derivadas de pacientes con cualquiera de las formas de EGC, se ha restaurado usando vectores retrovirales que contienen el cADN correspondiente al gen defectuoso.39–41 Otros estudios han utilizado líneas celulares mieloides llamadas PLB–985 como modelo in vitro de X–EGC, las cuales requieren la expresión de gp91phox para generar el anión superóxido. Una observación importante derivada de estos trabajos es que la expresión de pequeñas cantidades de gp91phox recombinante es suficiente para obtener una alta producción de anión superóxido, lo que sugiere que la proteína silvestre se encuentra naturalmente en exceso.42 Otro modelo celular utilizado en este campo de investigación es el de las células progenitoras de la médula ósea obtenidas de pacientes con EGC que luego son transducidas con diferentes tipos de vectores para después evaluar las células diferenciadas in vitro (granulocitos y monocitos), por medio de ensayos funcionales que permiten determinar la reconstitución del sistema NADPH oxidasa.17–19,43,44
Los estudios de transferencia genética retroviral utilizando células progenitoras mieloides obtenidas de la sangre periférica de pacientes con EGC han arrojado resultados satisfactorios. Por ejemplo, en un estudio preclínico se usó un vector bicistrónico retroviral derivado del virus de células madre murinas (MSCV, por su sigla en inglés), que brinda la posibilidad de evaluar clínicamente las células transducidas, ya que coexpresa el gen terapéutico de gp91phox y un gen marcador. Los resultados de este estudio mostraron hasta un 80% de transducción en las células progenitoras de la médula ósea CD34+ y entre los fagocitos diferenciados se alcanzó un 70% de los niveles normales de producción de anión superóxido.45
El estudio de la terapia génica en ratones Knockout que presentan EGC–X, ha permitido demostrar la eficiencia de los vectores basados en MSCV en la reconstitución de la actividad oxidasa. En este caso las células progenitoras hematopoyéticas provenientes de los ratones Knockout son transducidas con el gen CYBB, ubicado en el vector MSVC y luego estas células son trasplantadas a ratones singénicos tratados previamente con dosis mieloablativas de irradiación. La actividad NADPH oxidasa en neutrófilos y macrófagos, evaluada después del trasplante, muestra un aumento en la intensidad de expresión y en el porcentaje de células que expresan la gp91phox .46 Se han hecho estudios similares en modelos murinos con deficiencia de p47phox, utilizando células transducidas con vectores de tipo MFG–S que contienen el cADN que codifica la p47phox. Estos estudios han arrojado resultados similares a los anteriores ya que la evaluación postrasplante de la producción de anión superóxido en células progenitoras hematopoyéticas transducidas, muestra un aumento en la actividad NADPH oxidasa. Además, la respuesta a microorganismos patógenos de ratones Knockout tratados con terapia génica en comparación con ratones sin dicho tratamiento y bajo las mismas condiciones, permitió concluir que los niveles de bacteremia de los ratones con la corrección del defecto genético fueron significativamente más bajos que los de los controles.47
En los modelos murinos de EGC–X se ha restaurado parcialmente la actividad oxidasa hasta en un 50% de los neutrófilos circulantes en la sangre periférica, lo cual se refleja en la disminución satisfactoria de la neumonía por A. fumigatus.46 Además, en muestras biológicas quiméricas que contienen células murinas transducidas y sin transducir, se ha observado que 10% de los neutrófilos corregidos es un nivel suficiente para prevenir la infección respiratoria por dicho microorganismo.48 Estos resultados son prometedores si se tiene en cuenta que las mujeres portadoras del defecto EGC ligado al cromosoma X requieren solo el 5 al 10% de los neutrófilos circulantes en sangre periférica con actividad oxidasa normal para controlar las manifestaciones clínicas de la enfermedad. Es decir, que las células con corrección genética pueden alcanzar estos porcentajes de actividad oxidasa para lograr la resolución de los síntomas.18,46,48
Ensayos clínicos de la capacidad correctora
La capacidad correctora de la transferencia génica mediada por retrovirus también se ha evaluado en ensayos clínicos de fase uno de investigación. Para tal efecto se han utilizado como blanco de la terapia células CD34+ movilizadas hacia la sangre periférica. Uno de estos estudios se hizo en cinco pacientes con deficiencia de p47phox, de quienes se obtuvieron células CD34+ movilizadas a la sangre periférica, a las cuales se les introdujo un vector retroviral que contenía el cADN de p47phox, para luego reinfundirlas a los pacientes. En este ensayo se restauró la actividad oxidasa entre 6 y 29% en células diferenciadas in vitro a granulocitos, que fueron obtenidas a partir de las células CD34+ transducidas; sin embargo, se presentó un bajo porcentaje de neutrófilos corregidos en la sangre periférica, entre 0.004 y 0.05%, después de la transfusión de dichas células en los pacientes.49
Este resultado refleja una baja eficiencia de la transferencia génica mediada por retrovirus en células progenitoras hematopoyéticas humanas. Es interesante observar que en estudios realizados con modelos animales bajo las mismas condiciones y con el mismo protocolo, se obtuvo un 5% de células de la sangre periférica que contenían el provirus después del trasplante, lo que indica que la falla radica en la optimización de los procedimientos de terapia génica aplicados en células humanas.18,47,49 En otro ensayo clínico, células CD34+ fueron transducidas con retrovirus que transportaban el gen CYBB mediante el método de infección con fragmentos de fibronectina (CH–296) y en presencia de una combinación de citoquinas como el ligando 3 de la tirosina–quinasa fetal (FLT–3L, por su sigla en inglés), el factor de células madre (SCF, por su sigla en inglés) la trombopoyetina y el factor estimulante de colonias de granulocitos (Ì–CSF, por su sigla en inglés). Cuatro días después de la transducción, las células fueron reinfundidas al paciente y se encontraron neutrófilos con la corrección del defecto de la actividad oxidasa cuatro semanas después de cada ciclo de infusión, pero el número de estas células disminuyó con el paso del tiempo.26,28,50
Desventajas y correctivos
Una desventaja, tanto de la terapia génica como del trasplante alogénico de médula ósea, es la aplicación de condiciones mieloablativas a los individuos receptores antes del trasplante, con el fin de mejorar la implantación de las células que serán reinfundidas. Al respecto, en algunos estudios se han evaluado los procedimientos de transferencia génica en ensayos clínicos, en los cuales se reducen las dosis de radiación, a las que se exponen los pacientes que se van a trasplantar. Además, se ha utilizado el 5–fluorouracilo (5–FU), que bloquea de novo la síntesis de ADN y de ARN sin causar daños celulares irreversibles como sí lo hacen la radiación o los agentes utilizados en quimioterapia.22,51 Estos trabajos indican también que la infusión de niveles altos de células del donante, asociada con bajas dosis de radiación o con un régimen previo al trasplante basado en la administración de 5–FU, confiere niveles bajos pero estables de neutrófilos corregidos genéticamente in vivo.52 Además estos estudios preliminares también muestran una reducción parcial de la inflamación y de la formación de granulomas cuando los ratones en estudio fueron expuestos a hifas esterilizadas de Aspergillus fumigatus.18, 22, 51, 52
Vectores lentivirales
A pesar de que los vectores retrovirales han sido los más estudiados durante la década del desarrollo de la terapia génica, la alternativa actual son los vectores lentivirales. Los estudios reportados que utilizan lentivirus modificados para transportar insertos genéticos se basan en ensayos in vitro con líneas celulares como modelo de EGC, que se realizan bajo condiciones óptimas de transducción y aplicando técnicas que ofrecen vectores con muchas características de seguridad.27,53 También se han reportado estudios en modelos murinos NOD/SCID que reciben el trasplante de células CD34+ humanas transducidas, los cuales presentan corrección del defecto EGC–X. Los resultados que arrojan estos estudios indican que los vectores lentivirales muestran una mayor eficiencia de transducción en ensayos in vivo comparados con la eficiencia de transducción de los vectores retrovirales, los cuales resultan más eficientes que los lentivirales en ensayos ex vivo.54 Sin embargo, la intensidad de expresión por célula de la proteína de interés fue mejor en las células transducidas con retrovirus, que en las que contenían el vector lentiviral; este inconveniente puede superarse modificando el promotor en el constructo del vector lentiviral para lograr el aumento de la expresión génica. Además fue interesante observar que en los ensayos in vitro, las células transducidas con el vector lentiviral mostraron porcentajes de producción de anión superóxido y de expresión génica, muy similares a los resultados arrojados por las células de individuos normales, aunque mucho menores que los porcentajes que mostraron las células transducidas con el vector retroviral.27,53–55
CONCLUSIONES
La terapia génica se perfila como un tratamiento curativo prometedor para las enfermedades hereditarias hematopoyéticas. Para aplicarla en el ámbito clínico como tratamiento de la EGC, es indispensable desarrollar protocolos de transferencia génica más eficientes en los que se mejoren aspectos como: la construcción de vectores con características de seguridad de última generación y con poca capacidad de inducir respuesta inmune en el individuo receptor; la viabilidad de las células blanco en cultivo y las condiciones microambientales necesarias para una óptima transducción. Las únicas medidas implementadas para enfrentar los efectos adversos de la terapia génica son el análisis de riesgo contra beneficio en cada paciente y el seguimiento postratamiento; así se confirman los efectos terapéuticos esperados y se garantiza la ausencia de signos clínicos que indiquen la generación de mutagénesis insercional o la producción de virus competentes para la replicación.
Las células hematopoyéticas modificadas genéticamente podrían ser expandidas por medio de la selección positiva in vivo, lo que se refiere a la inclusión de un gen que confiera resistencia a un tratamiento mielotóxico en el constructo del vector y que permita la proliferación de las células que contengan dicho vector en un medio selectivo. Por último, la reducción de los requerimientos mieloablativos que preceden al trasplante, asociada con la infusión de un número mayor de células transducidas, puede redundar en una disminución del daño al estado inmunológico de los pacientes sin afectar la adherencia de las células trasplantadas.
REFERENCIAS BIBLIOGRÁFICAS
1. BABIOR BM, LAMBETH JD, NAUSEEF W. The neutrophil NADPH oxidase. Arch Biochem Biophys 2002; 397: 342–344. [ Links ]
2. VIGNAIS PV. The superoxide–generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci 2002; 59:1.428–1.459. [ Links ]
3. PATIÑO PJ. Enfermedad granulomatosa crónica y sistema NADPH oxidasa de las células fagocíticas: Una relación apasionante. Revista Colombiana de Inmunoalergia 1998; 7: 23–24. [ Links ]
4. HEYWORTH PG, CROSS AR, CURNUTTE JT. Chronic granulomatous disease. Curr Opin Immunol 2003; 15: 578–584. [ Links ]
5. ZAMBRANO E, ESPER F, ROSENBERG R, KIM J, REYES–MUGICA M. Chronic Granulomatous Disease. Pediatr Dev Pathol 2003; 6: 577–581. [ Links ]
6. GOLDBLATT D, THRASHER AJ. Chronic granulomatous disease. Clin Exp Immunol 2000; 122: 1–9. [ Links ]
7. JOHNSTON RB JR. Clinical aspects of chronic granulomatous disease. Curr Opin Hematol 2001; 8: 17–22. [ Links ]
8. PETERSEN JE, HIRAN TS, GOEBEL WS, JOHNSON C, MURPHY RC, AZMI FH, et al. Enhanced cutaneous inflammatory reactions to Aspergillus fumigates in a murine model of chronic granulomatous disease. J Invest Dermatol 2002; 118: 424–429. [ Links ]
9. MONTOYA CJ, AYALA A. Enfermedad granulomatosa crónica ligada al cromosoma X: 4 casos familiares fatales. Rev Asoc Colomb Alerg Asma Inmunol 1999; 8: 59–61. [ Links ]
10. PATIÑO PJ, PÉREZ JE, LÓPEZ JA, CONDINO–NETO A, GRUMACH A, BOTERO JH, et al. Molecular analysis of chronic granulomatous disease produced by defect in gp91 phox. Human Mutation 1999; 13: 29–37. [ Links ]
11. LÓPEZ JA, PATIÑO PJ, GARCÍA DE O D. Caracterización molecular en pacientes con Enfermedad Granulomatosa Crónica por deficiencia en p47 phox. Iatreia 1998; 11: 16– 21. [ Links ]
12. PATINO PJ, RAE J, NOACK D, ERICKSON R, DING J, DE OLARTE DG, et al. Molecular characterization of autosomal recessive chronic granulomatous disease caused by a defect of the nicotinamide adenine dinucleotide phosphate (reduced form) oxidase component p67–phox. Blood 1999; 94: 2.505–2.514. [ Links ]
13. MONTOYA CJ, SALGADO H, HENAO J, ORREGO JC, PATIÑO PJ. Guía de estudio y manejo del paciente sospechoso de alteraciones en la inmunidad mediada por las células fagocíticas. Rev Asoc Colomb Alerg Asma Inmunol 1999; 8: 23–26. [ Links ]
14. GARCÍA DE O D, PATIÑO PJ, SALGADO H, LÓPEZ JA, MONTOYA CJ, PÉREZ JE. Evaluación del paciente con inmunodeficiencia. Síndrome de Infección Recurrente Patológica. Medicina y Laboratorio 1997;7: 545–575. [ Links ]
15. PATIÑO PJ. Avances terapéuticos en las inmunodeficiencias primarias. Revista Colombiana de Inmunoalergia 1999; 8:14–18. [ Links ]
16. VALENZUELA CY. Ética científica de la terapia génica de individuos: Urgencia de la Cirugía Génica del ADN. Rev Méd Chile 2003;131:1.208–1.214. [ Links ]
17. MALECH HL. Progress in gene therapy for chronic granulomatous disease. J Infect Dis 1999;179 (Suppl 2): S318–25. [ Links ]
18. KUME A, DINAUER MC. Gene therapy for chronic granulomatous disease. J Lab Clin Med 2000; 135: 122–128. [ Links ]
19. GREZ M, BECKER S, SAULNIER S, KNOSS H, OTT MG, MAURER A, et al. Gene therapy of chronic granulomatous disease. Bone Marrow Transplant 2000; 25 (Suppl 2): S99–104. [ Links ]
20. CANDOTTI F. Gene therapy for immunodeficiency. Curr Allergy Asthma Rep 2001; 1: 407–415. [ Links ]
21. OTSU M, WADA T, CANDOTTI F. Gene therapy for primary immune deficiencies. Curr Opin Allergy Clin Immunol 2001; 1: 497–501. [ Links ]
22. SORRENTINO BP, NIENHUIS AW. The hematopoietic system as a target for gene therapy. In: Friedmann T, editor. The Development of the Human Gene Therapy. 1ª ed.New York: Cold Spring Harbor Laboratory Press; 1999. p. 351–426. [ Links ]
23. DUNBAR CE, WU T. Gene therapy for hematological disorders. In: Kresina TF, editor. An Introduction to Molecular Medicine and Gene Therapy. New York: Wiley–Liss; 2001. p. 133–151. [ Links ]
24. MORITZ T, WILLIAMS DA. Gene transfer into the hematopoietic system. Curr Opin Hematol 1994;1: 423–428. [ Links ]
25. COSSET FL, RUSSELL SJ. Targeting retrovirus entry. Gene Ther 1996; 3: 946–956. [ Links ]
26. KUME A, HANAZONO Y, MIZUKAMI H, URABE M, OZAWA K. Hematopoietic stem cell gene therapy: a current overview. Int J Hematol 1999; 69: 227–233. [ Links ]
27. PONDER KP. Vectors in gene therapy. In: Kresina TF, editor. An Introduction to Molecular Medicine and Gene Therapy. New York: Wiley–Liss; 2001. p. 77–111. [ Links ]
28. USMANI BA, FASSATI A, DICKSON G. Development of RNA virus for gene delivery. In: Cann AJ, editor. RNA Viruses: a Practical Approach. New York: Oxford University Press; 2000. p. 229–257. [ Links ]
29. FRIEDMANN T, YEE JK. Pseudotyped retroviral vectors for studies of human gene therapy. Nat Med 1995; 1: 275–277. [ Links ]
30. SALMONS B, GUNZBURG WH. Targeting of retroviral vectors for gene therapy. Hum Gene Ther 1993; 4: 129–141. [ Links ]
31. HACEIN–BEY–ABINA S, VON KALLE C, SCHMIDT M, MCCORMACK MP, WULFFRAAT N, LEBOULCH P, et al. LMO2–associated clonal T cell proliferation in two patients after gene therapy for SCID–X1. Science 2003; 302: 415–419. [ Links ]
32. GUNTER KC, KHAN AS, NOGUCHI PD. The safety of retroviral vectors. Hum Gene Ther 1993; 4: 643–645. [ Links ]
33. FRIEDMANN T. Gene therapy's new era: a balance of unequivocal benefit and unequivocal harm. Mol Ther 2003; 8: 5–7. [ Links ]
34. WOLFF JA. The 'grand' problem of synthetic delivery. Nat Biotechnol 2002; 20: 768–769. [ Links ]
35. FRIEDMANN T. Medical ethics. Principles for human gene therapy studies. Science 2000; 287: 2.163–2.165. [ Links ]
36. GOEBEL WS, DINAUER MC. Gene therapy for chronic granulomatous disease. Acta Haematol 2003; 110: 86–92. [ Links ]
37. LI F, LINTON GF, SEKHSARIA S, WHITING–THEOBALD N, KATKIN JP, GALLIN JI, et al. CD34+ peripheral blood progenitors as a target for genetic correction of the two flavocytochrome b558 defective forms of chronic granulomatous disease. Blood 1994; 84: 53–58. [ Links ]
38. SEKHSARIA S, GALLIN JI, LINTON GF, MALLORY RM, MULLIGAN RC, MALECH HL. Peripheral blood progenitors as a target for genetic correction of p47phox deficient chronic granulomatous disease. Proc Natl Acad Sci USA 1993; 90:7.446–7.450. [ Links ]
39. VOLPP BD, LIN Y. In vitro molecular reconstitution of the respiratory burst in B lymphoblasts from p47–phox–deficient chronic granulomatous disease. J Clin Invest 1993; 91: 201–207. [ Links ]
40. MALY FE, SCHUERER–MALY CC, QUILLIAM L, COCHRANE CG, NEWBURGER PE, CURNUTTE JT, et al. Restitution of superoxide generation in autosomal cytochrome–negative chronic granulomatous disease (A22(0) CGD)–derived B lymphocyte cell lines by transfection with p22phax cDNA. J Exp Med 1993; 178: 2.047– 2.053. [ Links ]
41. PORTER CD, PARKAR MH, COLLINS MK, LEVINSKY RJ, KINNON C. Efficient retroviral transduction of human bone marrow progenitor and long–term culture– initiating cells: partial reconstitution of cells from patients with X–linked chronic granulomatous disease by gp91–phox expression. Blood 1996; 87: 3.722–3.730. [ Links ]
42. ZHEN L, KING AA, XIAO Y, CHANOCK SJ, ORKIN SH, DINAUER MC. Gene targeting of X chromosomelinked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc Natl Acad Sci USA 1993; 90: 9.832–9.836. [ Links ]
43. DING C, KUME A, BJORGVINSDOTTIR H, HAWLEY RG, PECH N, DINAUER MC. High–level reconstitution of respiratory burst activity in a human X–linked chronic granulomatous disease (X–CGD) cell line and correction of murine X–CGD bone marrow cells by retroviral–mediated gene transfer of human gp91phox. Blood 1996; 88: 1.834 1.840. [ Links ]
44. KUME A, DINAUER MC. Retrovirus–mediated reconstitution of respiratory burst activity in X–linked chronic granulomatous disease cells. Blood 1994; 84: 3.311–3.316. [ Links ]
45. BARESE CN, GOEBEL WS, DINAUER MC. Gene therapy for chronic granulomatous disease. Expert Opin Biol Ther 2004; 4:1.423–1.434. [ Links ]
46. DINAUER MC, LI LL, BJORGVINSDOTTIR H, DING C, PECH N. Long–term correction of phagocyte NADPH oxidase activity by retroviral–mediated gene transfer in murine X linked chronic granulomatous disease. Blood 1999; 94: 914–922. [ Links ]
47. MARDINEY M, JACKSON SH, SPRATT SK, LI F, HOLLAND SM, MALECH HL. Enhanced host defense after gene transfer in the murine p47phox–deficient model of chronic granulomatous disease. Blood 1997; 89: 2.268–2.275. [ Links ]
48. BJORGVINSDOTTIR H, DING C, PECH N, GIFFORD MA, LI LL, DINAUER MC. Retroviral mediated gene transfer of gp91phox into bone marrow cells rescues defect in host defense against Aspergillus fumigatus in murine X–linked chronic granulomatous disease. Blood 1997; 89: 41–48. [ Links ]
49. MALECH HL, MAPLES PB, WHITING–THEOBALD N, LINTON GF, SEKHSARIA S, VOWELLS SJ, et al. Prolonged production of NADPH oxidase–corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci USA 1997; 94: 12.133–12.118. [ Links ]
50. ZIELSKE SP, GERSON SL. Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol Ther 2003; 7: 325–333. [ Links ]
51. YEE JK. Retroviral vectors. In: FRIEDMANN T, editor. The Development of Human Gene Therapy. New York: Cold Spring Harbor Laboratory Press; 1999. p. 21–45. [ Links ]
52. GOEBEL WS, DINAUER MC. Retroviral–mediated gene transfer and nonmyeloablative conditioning: studies in a murine X–linked chronic granulomatous disease model. J Pediatr Hematol Oncol 2002; 24:787–790. [ Links ]
53. NALDINI L. Lentiviral Vectors. In: Friedmann T, editor. The Development of the Human Gene Therapy. New York: Cold Spring Harbor Laboratory Press; 1999. p. 47–60. [ Links ]
54. ROESLER J, BRENNER S, BUKOVSKY AA, WHITINGTHEOBALD N, DULL T, KELLY M, et al. Third–generation, self–inactivating gp91(phox) lentivector corrects the oxidase defect in NOD/SCID mouse–repopulating peripheral blood–mobilized CD34+ cells from patients with X–linked chronic granulomatous disease. Blood 2002; 100: 4.381–4.390. [ Links ]
55. FRIEDMANN T. Changing roles for academia and industry in genetics and gene therapy. Mol Ther 2000;1: 9–11. [ Links ]
Recibido: 22 de agosto de 2005
Aceptado: 29 de agosto de 2005

