










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.19 no.1 Medellín Jan./Mar. 2006
ARTÍCULO DE REVISIÓN
Los linfocitos: modelo de estudio en las enfermedades de Alzheimer y Parkinson
Lymphocytes as a cell model to study Alzheimer's and Parkinson's diseases
Marlene Jiménez Del Rio1; Carlos Vélez–Pardo1
1. Profesores Asociados, Facultad de Medicina, Departamento de Medicina Interna, Grupo de Neurociencias de Antioquia, Universidad de Antioquia, Medellín, Colombia
Este artículo fue financiado por el 'Comité para el Desarrollo & la Investigación–CODI–UdeA' proyecto #9805, 9835, 9961; Colciencias #1115–04–10231; y 'Proyecto de Investigación en Enfermedades Neurodegenerativas' #8780.
RESUMEN
LAS ENFERMEDADES DE ALZHEIMER FAMILIAR (EAF) y de Parkinson familiar (EPF) son dos entidades neurodegenerativas progresivas que afectan a un grupo numeroso de individuos en el departamento de Antioquia, Colombia.
Hasta el presente no existe un tratamiento eficaz y definitivo que reduzca o detenga el deterioro clinicopatológico que padecen los pacientes de estas enfermedades.
Por estas razones, los procesos bioquímicos comprometidos en la pérdida neuronal han sido esencialmente estudiados en modelos biológicos. Los autores de este artículo, basados en su propia experiencia y en la de otros investigadores, postulan a los linfocitos como modelo celular de estudio para dilucidar las señales moleculares intracelulares activadas por el estrés oxidativo (EO) en las enfermedades de Alzheimer y Parkinson. Sorprendentemente, los linfocitos poseen una similitud celular y bioquímica con las células nerviosas. De hecho, los linfocitos expresan por lo menos seis sistemas biológicos, considerados como pertenecientes únicamente a las neuronas, tales como los sistemas catecolaminérgico, serotonérgico, acetilcolinérgico, glutamaérgico, noradrenérgico, gabaérgico. Debido a que los linfocitos son células con ciclo celular en la etapa G0, y presentan los sistemas metabólicos de síntesis, transporte y procesamiento de las proteínas precursora del beta–amiloide (APP), presenilinas y parkina, estas células constituyen un modelo celular ideal que permitirá una mejor comprensión de la señalización patológica de los procesos neurodegenerativos, y además permitirá realizar un enfoque racional de los diseños terapéuticos que detengan las causas de deterioro neuronal en los pacientes que padecen estas enfermedades.
PALABRAS CLAVE: ALZHEIMER, APOPTOSIS, LINFOCITOS, MODELO, PARKINSON
SUMMARY
ALZHEIMERS DISEASE (AD) AND PARKINSONS disease (PD) are two common progressive neurodegenerative disorders that affect a large group of individuals in Antioquia, Colombia. Up till now, there is not an effective and definitive therapeutic treatment aimed to reduce or retard the clinic and pathologic symptoms induced by both devastating diseases. Thus, the biochemical mechanism(s) involved in neural loss has mainly been studied in biological models. In this article the authors, based on their own scientific experience and others, have proposed lymphocytes as a cellular model to study the molecular signalling mechanism induced by oxidative stress in AD and PD. Remarkably, lymphocytes and neurons are cellularly and biochemically alike.
Indeed, lymphocytes express at least six different biological systems such as catecholaminergic, serotonergic, acetylcholinergic, glutamatergic, noradrenergic and GABAergic systems considered to be exclusively pertain to neural cells!. Given that lymphocytes are post–mitotic cells (i.e. G0 cell cycle) and possess the synthesis, transport and processing systems of amyloid–beta precursor protein (AβPP), presenilins and parkin proteins, these cells constitute not only an ideal bio–model to better understanding of the pathological signalling in those neurodegenerative processes, but also represent a meaningful tool to design innovative and rational therapeutic strategies focussed to arrest the neuronal death in Alzheimers and Parkinsons patients from Antioquia.
KEYWORDS: ALZHEIMER, APOPTOSIS, LYMPHOCYTES, MODEL, OXIDATIVE STRESS, PARKINSON
INTRODUCCIÓN
LA ENFERMEDAD DE ALZHEIMER FAMILIAR (EAF) y la enfermedad de Parkinson familiar (EPF) son dos entidades neurodegenerativas progresivas que afectan a un grupo numeroso de individuos en el departamento de Antioquia, Colombia.1,2 Clínicamente, la EAF se caracteriza por presentar un deterioro intelectual progresivo que involucra no solamente la pérdida de las funciones de memoria, orientación y lenguaje, sino que también trastorna otros componentes funcionales más complejos como la personalidad, el juicio, la solución de problemas, la capacidad de cálculo, la habilidad en construcciones visuoespaciales, que finalmente induce un estado absoluto de dependencia mental y física.3
Patológicamente, la EAF se caracteriza por atrofia cerebral severa y por la presencia de cinco marcadores típicos: una marcada reactividad de la glía, despoblamiento neuronal, excesivo depósito de placas neuríticas (PN), de ovillos neurofibrilares (ONF), y de hierro.4,5 Notoriamente, el principal componente de las PN es un fragmento proteico denominado beta–amiloide (Aβ [ 1–42 ] ). Este péptido se genera a partir del procesamiento proteolítico de la proteína precursora de β–amiloide (PPA) de 695/771 amino ácidos por la acción enzimática combinada de la β aspartil proteasa de membrana denominada BACE–1, (por su sigla en inglés β–site APP–cleaving enzyme), γ secretasas (complejo multiproteico compuesto de la proteína presenilina (PS), nicastrina (Nct), APH–1 (por su sigla en inglés anterior pharynx–defective phenotype), PEN–2 (por su sigla en inglés PSenhancer), las cuales generan además del fragmento Aβ1–42 amiloidogénico, un fragmento soluble PPAsβ y un dominio intracelular AICD (por su sigla en inglés APP intracellular domain) de función desconocida. Por otra parte, si la PPA es proteolizada por la acción combinada de las enzimas α(de la familia de metaloproteasas ADAM, por su sigla en inglés a disintigrin and metalloproteases) γ secretasas, no se produce el fragmento patogénico Aβ1–42, sino que se generan tres fragmentos: uno soluble PPAsα, un péptido p3 y el dominio intracelular AICD.6,7
Hasta el presente, tres genes causales (presenilina–1 [ PS–1 ], presenilina–2 [ PS–2 ], APP) localizados en los cromosomas 14, 1, 21, respectivamente, y un gen de riesgo (apolipoproteína E, APOE) localizado en el cromosoma 22, han sido asociados con un depósito exagerado de Aβ [ 1–42 ] en el parénquima cerebral y en la vasculatura (angiopatía cerebral) en la EAF. Sorprendentemente, se han identificado 177 mutaciones en los tres genes8 (PS–2: 11 mutaciones; APP: 25 mutaciones; PS–1: 141mutaciones), de las cuales resulta la mutación en el exón 8 (cambio en el nucleótido 839 de una adenina por una citosina, gAa>gCa) de la PS–1,9 y que codifica para la proteína (467 amino ácidos) mutada E280A (resultado de un cambio del residuo de ácido glutámico por una alanina en la posición 280). Infortunadamente, hasta el momento, el grupo familiar más numeroso del mundo con una mutación puntual en el gen de la PS–1 ha sido descrito en Colombia.1,9
Efectivamente, se ha demostrado que la mutación E280A induce un incremento exagerado en la producción y acúmulo del Aβ [ 1–42 ] en el parénquima cerebral y en las paredes de los vasos sanguíneos, resultando en una lesión cerebral grave que lleva finalmente a un inicio clínico temprano o precoz (menos de 60 años) de la enfermedad.10–14 Como resultado de estos cambios genético–moleculares, se prevé un aumento alarmante de nuevos casos de EAF en los próximos años. Por otra parte, la EPF se manifiesta en Antioquia como una enfermedad de Parkinson juvenil autosómica recesiva (EPJAR),2 y se caracteriza clínicamente por la manifestación de movimientos lentos del cuerpo, rigidez muscular, temblor, pérdida del balance en la postura, y por su aparición temprana en sujetos menores de 40 años.15 Patológicamente, se caracteriza por una degeneración selectiva de las neuronas dopaminérgicas de la región pars compacta de la sustancia negra y por la presencia de abundantes depósitos de hierro.16 A diferencia de la EP esporádica, los pacientes que padecen EPJAR responden al tratamiento con L–DOPA, no presentan las típicas inclusiones eosinofilicas neurofilamentosas compuestas principalmente por la proteína α–sinucleína, denominados cuerpos de Lewis, y genéticamente se caracterizan por presentar una mutación en el gen de la parkina–2, que resulta de un cambio de una cisteína por una tirosina. Esta mutación es conocida como mutación paisa parkina–C212Y.2,17
Estudios clínicos, patológicos y genéticos han aportado información importante para el conocimiento de estas dos entidades neurológicas,1,2,10–14,17 pero aún no se ha logrado establecer claramente cuáles son los mecanismos moleculares y patológicos de señalización celular que desencadenan la pérdida neuronal en el cerebro de los pacientes con EAF, EPF, o en los que tienen manifestaciones clinicopatológicas de ambas enfermedades. Además, aunque los pacientes de Alzheimer y Parkinson reciben tratamientos que mejoran sus manifestaciones clínicas, hasta el presente no existe una terapia eficaz y definitiva que reduzca o detenga el deterioro clinicopatológico que padecen los pacientes de estas enfermedades neurodegenerativas. Por estas razones, los procesos bioquímicos comprometidos en la muerte neuronal han sido esencialmente estudiados en modelos animales, en células primarias neuronales o en líneas celulares de origen neuronal o no neuronal. Por lo anterior, es imperativo responder a dos preguntas críticas: 1) ¿Cuáles son los mecanismos moleculares que inducen la muerte neuronal selectiva en las enfermedades de Alzheimer y Parkinson? 2) ¿Qué estrategias terapéuticas moleculares tempranas podrían implementarse para prevenir, retardar o interrumpir el impacto patológico en estas enfermedades?
UNA HIPÓTESIS + UN MODELO = UNA EXPLICACIÓN
Para responder a estas dos preguntas, la investigación científica dispone de dos procedimientos básicos: 1) Planteamiento de una hipótesis del fenómeno que se va a estudiar. De hecho, las hipótesis expresan lo que estamos buscando o tratando de probar y se definen como explicaciones tentativas del fenómeno investigado, formuladas a manera de proposiciones que relacionan dos o más variables (v. gr. EO y enfermedades neurodegenerativas) que se apoyan en conocimientos organizados y sistematizados y que deben ser sujetas a comprobación empírica y verificación en la realidad. 2) Selección de un modelo (in vivo, in vitro o in situ) que nos permita comprobar, verificar o rechazar dichas suposiciones o hipótesis, como primera aproximación explicativa del objeto observado. Finalmente, la experimentación y la compilación rigurosa de observaciones, nos permitirá formular una teoría, la cual es una explicación conveniente y convincente del mundo observable, que será aceptada como un hecho empleando un modelo de estudio.
Por definición, un modelo es una descripción simplificada (¡pero no simplista!) de la realidad. Se utiliza para propósitos de predicción y control, y nos permite mejorar nuestra comprensión de las características del comportamiento de la realidad estudiada de una forma más selectiva que si se la observara directamente. Específicamente, entendemos un modelo biomédico como un organismo unicelular o pluricelular que pueda reemplazar y/o representar total o parcialmente los aspectos fisiológicos o patológicos del ser humano, con la capacidad de aportar nueva información para comprender el funcionamiento normal o anormal desde un gen hasta un fenotipo, y poder alcanzar el objetivo final que es una intervención preventiva o terapéutica en las enfermedades humanas. Nótese, sin embargo, que para ser seleccionado un modelo biomédico no necesariamente debe ser una réplica exacta de una condición o enfermedad humana.
Durante los últimos años se han propuesto varias hipótesis con el propósito de explicar las causas de la pérdida neuronal de la región hipocampal y de la sustancia negra en los pacientes con Alzheimer y Parkinson familiares, respectivamente. De hecho, la hipótesis de mayor impacto y aceptación postula que estas enfermedades neurodegenerativas resultan de un proceso de (EO), definido como un desequilibrio entre la producción excesiva de especies reactivas de oxígeno [ ERO ] tales como el anión superóxido [ O.2 ], el peróxido de hidrógeno [ H2O2 ], radicales de hidroxilo [ .OH ], y la disminución o ausencia de los sistemas celulares de respuesta antioxidante, sean enzimáticos o no enzimáticos, generado dicho EO por el péptido de Aβ1–42 en la EAF, o por alteraciones en la proteína parkina en la EPF, respectivamente. Es de anotar que, hasta el presente, no se han establecido completamente los mecanismos moleculares exactos que inducen la muerte neuronal en el sistema hipocampal, catecolaminérgico o serotonérgico. En este sentido, hemos presentado la hipótesis que en la EAF y la EPF intervienen cascadas de eventos de señalización moleculares y patológicos comunes inducidos por el EO.18 Por lo tanto, una terapia unificada podría beneficiar a los pacientes con Alzheimer y Parkinson. Con el fin de profundizar más en estos aspectos, nuestro grupo de investigación se ha propuesto los siguientes objetivos: 1) establecer los mecanismos moleculares de señalización de muerte inducidos por el EO generado por el dominio tóxico del Aβ[ 1–42 ], el fragmento Aβ[ 25–35 ], los metales (v. gr. hierro, cobre, manganeso, zinc), las monoaminas (dopamina, serotonina) y sus toxinas relacionadas (6–OHDA, 5,6 y 5,7–DHT); 2) establecer los mecanismos moleculares de señalización de supervivencia celular inducidos por moléculas de origen animal o vegetal (con capacidad antioxidante) contra el EO, para diseñar nuevas estrategias terapéuticas; 3) identificar mutaciones génicas (nuevas o establecidas) que puedan ser causales o modificadoras en los procesos de pérdida neuronal en los trastornos neurodegenerativos; 4) identificar biomarcadores de EO para realizar un diagnóstico precoz en los pacientes con EAF y EPF. Como modelo de estudio hemos seleccionado los linfocitos de sangre periférica (LSP) humana. Pero, ¿por qué los linfocitos?

LOS LINFOCITOS: UN MODELO NEURONAL
Si aceptamos que un modelo es por definición 'una descripción simplificada de la realidad , [ que ] nos permite mejorar nuestra comprensión de las características del comportamiento de la realidad estudiada de una forma más selectiva que si se la observara directamente , [ y que el modelo ] no necesariamente debe ser una réplica exacta de una condición o enfermedad humana para ser seleccionado ', este sería por sí solo un enfoque suficiente para dar una respuesta satisfactoria. Sin embargo, la selección de los linfocitos como modelo de estudio neuronal va más allá de la curiosidad o del simple gusto de los investigadores. De hecho, ¡nos deja sorprendidos la similitud celular y bioquímica de los linfocitos cuando se los compara con las neuronas! Por lo tanto, es clara por varias razones la justificación para postular los linfocitos como un modelo ideal de estudio del EO y la muerte celular en las enfermedades neurodegenerativas, y en especial de la EP. En primer lugar, los linfocitos y las neuronas presentan la misma maquinaria molecular que lleva a una morfología de muerte celular típica de apoptosis. Este tipo de muerte celular fue descrito por primera vez por Kerr y colaboradores en 1972,19 y ampliamente ilustrado utilizando linfocitos en 1995.20 En este último estudio se muestran claramente, utilizando la técnica de microscopía electrónica, las características que definen hasta el presente la apoptosis: condensación y fragmentación de la cromatina en masas granulares uniformes bien definidas, condensación del citoplasma con preservación estructural de sus organelos, vacuolización del citoplasma y formación de vesículas con fragmentos de núcleo u organelos, denominados cuerpos apoptóticos. Naturalmente, la maquinaria molecular responsable de esta morfología apoptótica hace parte del programa de muerte, de tal suerte que los linfocitos fueron posteriormente utilizados para definir las características bioquímicas de la apoptosis. Es decir, la vía de muerte extrínseca, o dependiente de receptores (v.gr. FasL) y la vía de muerte intrínseca, o dependiente de la señalización desencadenada por despolarización mitocondrial. De hecho, estas dos observaciones (morfológica y bioquímica) han sido esenciales para explicar y describir la muerte neuronal.21,22 En segundo lugar, las neuronas y los linfocitos son células postmitóticas en fase G0 en su ciclo celular. Es decir, estas células permanecen sin proliferar toda su vida funcional. En tercer lugar, aunque su origen embrionario es diferente –las neuronas provienen del ectodermo y los linfocitos, del mesodermo– se ha demostrado en estudios in vitro e in vivo que las neuronas pueden originarse por diferenciación de células madre mesenquimatosas provenientes de la médula ósea o de células madre hematopoyéticas.23,24 En cuarto lugar, ¡los linfocitos despliegan al menos seis sistemas biológicos, considerados como pertenecientes únicamente a las neuronas!, y comparten otros sistemas con similar función (véase tabla).
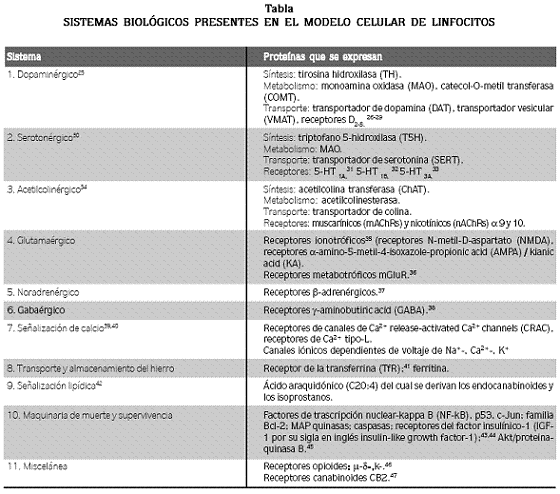
Finalmente, los linfocitos presentan los sistemas metabólicos de síntesis, transporte y procesamiento de la APP,48 expresan presenilinas,49 y expresan la proteína parkina.50 Adicionalmente, en los últimos años se han publicado aproximadamente 490 artículos científicos relacionados con diferentes aspectos de la biología celular y molecular de las enfermedades de Alzheimer y de Parkinson (búsqueda www.scopus.com, desde el año 1990 hasta el presente, 2005).
Por estas razones, consideramos que los linfocitos constituyen un excelente modelo para el estudio de los procesos moleculares de neurodegeneración inducidos por EO, extrapolables a condiciones neuropatológicas y comparables con investigaciones en modelos neuronales.
LOS LINFOCITOS: MECANISMOS DE SEÑALIZACIÓN MOLECULAR QUE EXPLICAN EL EO COMO AGENTE CAUSAL EN LAS ENFERMEDADES DE ALZHEIMER Y PARKINSON
En un trabajo previo publicado por Walkinshaw y Waters51 se demostró que la 6–OHDA inducía apoptosis en la línea neuronal PC–12. Este artículo, y una búsqueda cuidadosa en la literatura científica, nos indicaron que no existían reportes sobre los efectos tóxicos de la 6–OHDA, 5,6–DHT y 5,7–DHT en un modelo celular único. Por lo tanto, con el objetivo de ampliar el conocimiento con estos estímulos tóxicos in vitro, y aprovechando la posibilidad técnica de evaluar directamente la morfología celular normal y apoptótica con la tinción de viabilidad naranja de acridina/bromuro de etidio (NA/BE), inicialmente logramos evidenciar que concentraciones crecientes (50, 150, 250 µM) de las toxinas dopaminérgicas y serotonérgicas inducían apoptosis (10%, 50% y 78%, respectivamente, con 6–OHDA) en LSP. Este resultado nos condujo a determinar si el efecto apoptótico ocurría por un mecanismo específico intracelular y si este efecto era dependiente de los productos de oxidación (quinonas y ERO) de dichas toxinas. Efectivamente, empleando el inhibidor específico de transporte de monoaminas, la desipramina, y los antioxidantes como el ácido ascórbico, N–acetil–cisteína, 17β–estradiol, observamos una reducción de su efecto tóxico comparable al observado con LSP sin ningún tratamiento (<1% de índice apoptótico). Estos resultados claramente mostraron que la acción nociva de estos estímulos ocurría en el interior de la célula y que requería un proceso de oxidación.
Posteriormente, nos interesamos en investigar cuál era específicamente la especie reactiva de oxígeno generada durante el proceso de oxidación de estas neurotoxinas. De hecho, no fue una sorpresa encontrar que el H2O2 es la especie que mayoritariamente se genera en este proceso de oxidación, pero lo más interesante fue determinar cuál era la conexión entre el H2O2 y la morfología apoptótica. Para dar respuesta a esta pregunta realizamos cuatro experimentos básicos: 1) determinamos la cinética de producción de H2O2 por las tres toxinas y, simultáneamente, evaluamos la morfología de apoptosis; 2) inhibimos la acción de la proteasa caspasa–3, principal molécula ejecutora de muerte neuronal por apoptosis, utilizando el bloqueador específico Ac–DEVD–cho; 3) también bloqueamos la síntesis de proteínas y del mRNA, utilizando los inhibidores actinomicina–D y cicloheximida, respectivamente; 4) finalmente, utilizando la técnica de reconocimiento por immunohistoquímica, determinamos si los factores de transcripción como el factor nuclear kappa–B (por su sigla en inglés NF–κB), p53 y c–Jun estaban implicados como moléculas activas en el proceso de muerte programada. De acuerdo con este diseño experimental, logramos demostrar que la generación de H2O2 es concomitante con la morfología típica de apoptosis, y que esta depende de la activación de la caspasa–3.
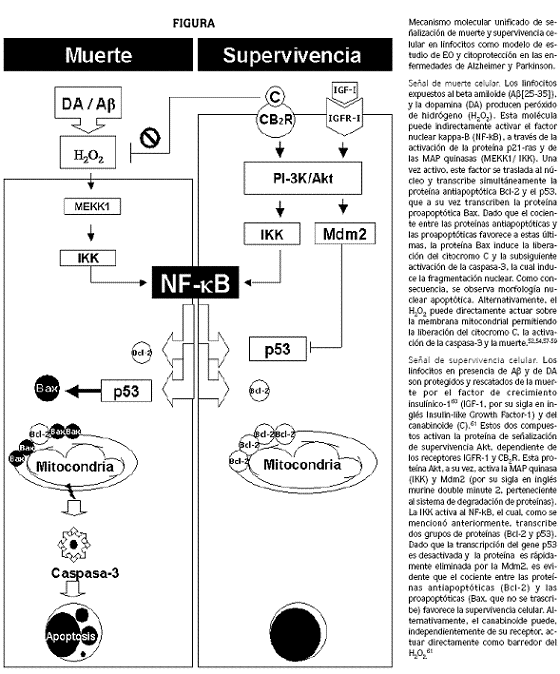
Además, logramos demostrar que la inducción apoptótica por parte de las toxinas/ (H2O2) dependía de la síntesis de novo de mRNA y proteínas. Importantemente, detectamos la activación simultánea de los factores NF–κB, p53, c–Jun asociada a la morfología apoptótica en linfocitos expuestos a las neurotoxinas. Tomados en conjunto, estos hallazgos nos permitieron concluir: primero, que la acción citotóxica, expresada en términos de porcentajes apoptóticos, de la 6–OHDA, la 5,6 y 5,7–DHT, y por consiguiente la generación del H2O2, dependían de la concentración y velocidad de oxidación de las toxinas; segundo, que el H2O2 es una molécula que desencadena una cascada de eventos secuenciales conducentes a apoptosis, a saber: [ 6–OHDA, 5,6–DHT; 5,7–DHT ]> quinonas + H2O2 >MAP quinasas> c–Jun, NF–κB > p53 > caspasa–3 > fragmentación nuclear del ADN. Finalmente, la dilucidación de los eventos moleculares comunes involucrados en la inducción de apoptosis por estas neurotoxinas, proporcionó nueva información para comprender la muerte neuronal en la EP, y sugirió que el EO juega un papel predominante en la patogénesis de la EP.52 Los resultados anteriores y la observación de Behl y colaboradores (1994)53 que el Aβ producía H2O2, nos incitaron a determinar si el Aβ[ 25–35 ] podría causar efectos similares a los observados con la 6–OHDA. Empleando técnicas de visualización de apoptosis, detección de H2O2, bloqueo de la síntesis de novo de mRNA y proteínas, inhibición específica de la caspasa–3, inhibición específica del NF–κB con el bloqueador PDTC, detección de la activación nuclear del NF–κB por las técnicas de reconocimiento immunohistoquímico y por el ensayo de cambio de movilidad electroforética (EMSA, sigla en inglés), detección con immunohistoquímica de la activación de p53 y c–Jun, logramos efectivamente demostrar que el péptido tóxico (10µM) AΒ[ 25–35 ] produce H2O2, y que a su vez, este compuesto induce una cascada específica de señalización de muerte celular análoga a la descrita con la 6–OHDA, es decir, [ Aβ(25–35) ] > H2O2 > MAP quinasas> c–Jun, NF–κB > p53 > caspasa–3 > fragmentación nuclear = apoptosis54 (véase figura, señal de muerte).
Es de anotar que estudios reportados por McLlelan et al., (2003)55 han corroborado nuestras observaciones. Esencialmente, estos investigadores han demostrado que las placas de Aβ in vivo en un modelo transgénico de Alzheimer y ex vivo a partir de tejido humano postmortem de Alzheimer eran capaces de producir radicales libres de oxígeno y H2O2.
Más aún, observaciones recientes en nuestro laboratorio mostraron la activación del NF–κB, p53, c–Jun y Par–4 en tejido postmortem (in situ) de pacientes con Alzheimer.56 Interesantemente, también demostramos que (25µM) Fe2+ incrementa de dos a cinco veces el efecto tóxico del Aβ en un intervalo de 24 a 48 horas. Este incremento apoptótico resultó de la propiedad química del Fe2+ de potenciar el Aβ para la generación de ERO y la producción de radicales libres de oxígeno. Es de anotar que el efecto nocivo del Aβ en presencia del Fe2+ fue independiente de la activación de NF–κB, p53 y c–Jun.54 Con estas observaciones logramos establecer un mecanismo operacional alternativo y modulado por el metal, en el cual, [ Aβ ] > H2O2 + Fe2+ > ERO > caspasa–3 > fragmentación nuclear = apoptosis. En conclusión, tomadas en su conjunto, estas investigaciones brindan una explicación del efecto nocivo de las neurotoxinas y del programa de activación molecular de muerte que se desencadena a partir de la molécula H2O2, en concordancia con las características típicas de la apoptosis. Es de resaltar que aunque la cascada de eventos moleculares inducida por las neurotoxinas y el Aβ presenta características bioquímicas comunes, no se había establecido una relación directa entre estos eventos moleculares (EO) y los factores genéticos. Este fue un objetivo que nos propusimos en la siguiente investigación.
Gracias a un estudio previo realizado por Pineda–Trujillo y colaboradores en Colombia (2001),2 en el cual se había reportado una nueva mutación consistente en la sustitución de una cisteína por una tirosina en el codón 212 en el gen parkina–2, en individuos provenientes de dos grupos familiares diagnosticados con la enfermedad de Parkinson juvenil autosómica recesiva (PJAR), nos interesamos en determinar la relación entre los factores genéticos y el impacto del EO en la PJAR. Con este propósito, y dado que los linfocitos expresan el isoformo 3 de la parkina, proteína comprometida en la regulación y degradación de las proteínas no plegadas resultantes de un proceso de estrés, seleccionamos 3 pacientes homocigotos recesivos (C212Y), un paciente heterocigoto (C212Y/C), y cuatro individuos normales (C212C). Efectivamente, logramos establecer que la mutación C212Y causa un aumento en la sensibilidad o susceptibilidad de los linfocitos de los pacientes homocigotos recesivos al EO generado por los estímulos tóxicos de H2O2, Fe2+ y DA, comparados con linfocitos de pacientes heterocigotos o normales. Adicionalmente, evidenciamos que la DA induce apoptosis por cuatro vías moleculares alternativas, convergentes en la activación de la caspasa–3: 1) una vía dependiente del NF–κB; 2) una vía dependiente de la alteración directa de la mitocondria por exposición al H2O2; 3) por exposición a radicales de hidroxilo (OH); y 4) por incremento de las proteínas no plegadas inducidas por estrés.57 Estos hallazgos muestran por primera vez que los factores genéticos, los ambientales y el Fe2+ contribuyen a la patogenia de la enfermedad de PJAR.
Curiosamente, el acúmulo de Fe2+ en el cerebro es una característica neuropatológica notable en la enfermedad de PJAR similar a la observada en la EAF y la EP. Evidentemente, esta característica sugiere que el Fe2+ juega un papel central en la patogénesis de estas enfermedades. Sin embargo, no se había dilucidado claramente si el Fe2+ constituye una causa primaria o secundaria en el deterioro neuronal en estas enfermedades; si la toxicidad del Fe2+ se debe a alteraciones en su metabolismo y/o cuál es su mecanismo de toxicidad. Por lo tanto, nos interesamos en investigar el mecanismo citotóxico del Fe2+ en el modelo de LSP. Adicionalmente, evaluamos otros metales tales como el Cu2+, Mn2+ y Zn2+ involucrados en otras enfermedades neurológicas. Con este propósito, expusimos LSP a concentraciones crecientes (50, 100, 250, 500, 1.000 µM) de estos metales. Usando el análisis morfológico con la tinción NA/BE, logramos observar que 500 µM de los metales provocan un porcentaje máximo de apoptosis (22–30%) y un porcentaje mínimo de necrosis (3–7%). Por otra parte, concentraciones menores de 500µM fueron inocuas, mientras que 1.000 µM provocaron principalmente necrosis (>40%). Estos resultados nos permitieron seleccionar la concentración de 500 µM para los siguientes experimentos. De hecho, esta concentración utilizada como estímulo deletéreo es comparable con los niveles de metales reportados in situ en las placas de los cerebros provenientes de pacientes con la EA (1mM Fe2+) y en la región de la sustancia negra de los cerebros de pacientes con la EP (13–15 µg de Fe2+/ por kilogramo de peso de tejido seco). Con este procedimiento experimental, evidenciamos por primera vez que los metales inducen apoptosis en LSP a través de la producción de H2O2 y radicales de hidroxilo (.OH), los cuales causan despolarización mitocondrial, activación de la caspasa–3, y fragmentación nuclear independientemente de la activación de los factores de trascripción NF–κB y p53.58 Estos datos sugieren efectivamente que los metales podrían constituir la causa primaria de los procesos neurodegenerativos en la EA y la EP, iniciada por una generación per se de H2O2 por los estados activos de óxido–reducción de los metales y OH formados por la reacción de Fenton (Fe2+ + H2O2 = Fe3+ + .OH + –OH). Estos hallazgos ilustran la importancia de los LSP como modelo de búsqueda para las estrategias antioxidantes que remuevan el H2O2 y el OH asociados a las reacciones catalizadas por metales en las enfermedades neurodegenerativas.
En resumen, estas investigaciones sugieren que las enfermedades de Alzheimer y Parkinson comparten un mecanismo común de señalización molecular de muerte neuronal y que el H2O2 es una molécula cardinal desencadenante del proceso neurodegenerativo. Actualmente, nuestra teoría titulada 'Mecanismo molecular unificado de EO en las enfermedades de Alzheimer y Parkinson', postula un posible escenario de los eventos fundamentales que llevan a apoptosis neuronal en estas enfermedades (véase figura).
Dado que hasta el momento no existen terapias efectivas contra estos trastornos neurológicos, se han investigado nuevas alternativas terapéuticas tales como el desarrollo de vacunas, la neuroprotección con factores de crecimiento y las moléculas derivadas de la botánica popular, entre otros. En este sentido, hemos demostrado que el 17β–estradiol57,59 y la testosterona57 protegen los linfocitos contra la muerte celular por apoptosis inducida por la combinación de dopamina y hierro. Recientemente hemos investigado el efecto citoprotector del factor de crecimiento insulínico–1 y los canabinoides como agentes moduladores en la señalización de supervivencia celular contra el EO estimulado por el Aβ(25–35), H2O2 y la dopamina.
En la literatura científica se reporta el IGF–1 y los canabinoides como moléculas neuroprotectoras contra el EO generado por el Aβ y el H2O2. Sin embargo, su mecanismo molecular de acción no se había establecido completamente. Aprovechando que los linfocitos expresan los receptores de IGF–1,43,44 conocidos como IGF–1R y que expresan el receptor canabinoide 2 (CB2),47 en condiciones experimentales análogas realizadas en trabajos previos, logramos demostrar claramente que el IGF–160 y los canabinoides61 desencadenan una cascada de eventos moleculares dependientes de sus receptores, que activan a su vez la proteína Akt/PκB, la cual está ligada a la activación (por fosforilación) de otras proteínas como el NF–κB (recordemos que en investigaciones previas habíamos asociado el factor NF–κB con apoptosis). ¿Cómo logra entonces este factor regular la decisión de muerte o supervivencia celular si transcribe simultáneamente señales evidentemente contradictorias, de muerte y de supervivencia?
Inicialmente pensamos que habría otras proteínas que interactuaban con el NF–κB, y que eran capaces de modular y/o regular su actividad transcriptora. Esta suposición resultó ser verdadera, y la respuesta a esta inquietud se dedujo de la observación que el NF–κB transcribe genes proapoptóticos y antiapoptóticos, los cuales, dependiendo de su relación numérica, deciden el destino de la célula. En otras palabras, si las proteínas proapoptóticas superan a las antiapoptóticas, el resultado será la muerte celular, y en el caso contrario el resultado será la supervivencia celular. Esta conclusión está sustentada por dos observaciones experimentales: en primer lugar, el factor NF–κB transcribe el gen p53, el cual a su vez transcribe genes proapoptóticos como Bax. En segundo lugar, la activación de la Akt/PκB no solo activa el NF–κB, sino que fosforila y activa la proteína Mdm2, la cual a su vez causa la eliminación de p53. De tal modo que esta última acción deja a las proteínas antiapoptóticas (Bcl–2) en cantidad numérica superior a las proteínas proapoptóticas (Bax), para evitar el colapso de la mitocondria y la muerte celular (véase figura, señal de supervivencia). No obstante, por procedimientos experimentales adicionales, logramos demostrar que los canabinoides no solo ejercen una acción citoprotectora contra el EO a través de la señalización del receptor CB2, sino que también son capaces de operar como moléculas antioxidantes, actuando directamente como barredores del H2O2.61
En conjunto, estas investigaciones demuestran que el uso de modelos simplificados, como los linfocitos, permite una mejor comprensión de la señalización patológica de los procesos neurodegenerativos, y además nos permitirá realizar una aproximación racional en los diseños terapéuticos que detengan o retarden de manera eficaz o definitiva las causas de deterioro neuronal en los pacientes que padecen las enfermedades de Alzheimer y Parkinson.
REFERENCIAS BIBLIOGRÁFICAS
1. LOPERA F, ARDILLA A, MARTINEZ A, MADRIGAL L, ARANGO–VIANA JC, LEMERE CA, et al. Clinical features of early–onset Alzheimer disease in a large kindred with an E280A presenilin–1 mutation. JAMA 1997; 277: 793–799. [ Links ]
2. PINEDA–TRUJILLO N, CARVAJAL–CARMONA LG, BURITICA O, MORENO S, URIBE C, PINEDA D, et al. A novel Cys212Tyr founder mutation in parkin and allelic heterogeneity of juvenile Parkinsonism in a population from North West Colombia. Neurosci Lett 2001; 298: 87–90. [ Links ]
3. REISBERG B, FRANSSEN EH. Clinical stages of Alzheimers disease. En: DE LEON MJ, ed. An atlas of Alzheimers disease. New York, London: The Parthenon Publishing Group; 1999:11–29. [ Links ]
4. ESIRI MM. The neuropathology of Alzheimers disease. En: DAWBARN D, ALLEN SJ, eds. Neurobiology of Alzheimers disease. Oxford: Oxford University Press; 2001: 33–53. [ Links ]
5. SMITH MA, HARRIS PL, SAYRE LM, PERRY G. Iron accumulation in Alzheimer disease is a source of redox–generated free radicals. Proc Natl Acad Sci USA 1997; 94: 9.866–9.868. [ Links ]
6. WILQUET V, DE STROOPER B. Amyloid–beta precursor protein processing in neurodegeneration. Curr Opin Neurobiol 2004; 14: 582–588. [ Links ]
7. HAASS C. Take five–BACE and the γ–secretase quartet conduct Alzheimers amyloid β–peptide generation. EMBO J 2004; 23: 483–488. [ Links ]
8. htpp://www.molgen.ua.ac.be/ADMutations/ [ Links ]
9. Alzheimer´s Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genetics 1995; 11: 219–222. [ Links ]
10. Pau P, ROE C, VILLEGAS CA, BEDOYA GJ, CHAKRAVERTY S, GARCÍA GP, et al. Apolipoprotein Ee4 modifies Alzheimers disease onset in an E280A PS1 kindred. Ann Neurol 2003; 54: 163–169. [ Links ]
11. ARANGO JC, IGLESIAS J, LOPERA FJ. Neuropsychological study of familial Alzheimers disease caused by mutation E280A in the Presenilin 1 gene. Am J Alzheimers Dis Other Demen 2003; 18: 137–146. [ Links ]
12. GÓMEZ T, GROWDON WB, MCNAMARA MJ, NOCHLIN D, BIRD TD, ARANGO JC, et al. The impact of different presenilin 1 and presenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimers disease brain. Brain 1999; 122: 1.709–1.719. [ Links ]
13. LEMERE CA, LOPERA F, KOSIK KS, LENDON CL, OSSA J, SAIDO TC, et al. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med 1996; 2:1.146–1.150. [ Links ]
14. JOHNSON KA, LOPERA F, JONES K, BECKER A, SPERLING R, HILSON J, et al. Presenilin–1–associated abnormalities in regional cerebral perfusion. Neurology 2001; 56: 1.545–1.551. [ Links ]
15. ZHANG Y, DAWSON VL, DAWSON TM. Parkin: clinical aspects and neurobiology. Clin Neurosci Res 2001; 1: 467–482. [ Links ]
16. TAKANASHI M, MOCHIZUCHI H, YOKOMIZO K, HATTORI N, MORI H, YAMAMURA Y, et al. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP). Parkinsonism Relat Disord 2001; 7: 311–314. [ Links ]
17. SANCHEZ JL, BURITICÁ O, PINEDA DA, URIBE CS, PALACIO LG. Prevalence of Parkinsons disease and Parkinsonism in a Colombian population using the capture–recapture method. Int J Neurosci 2004; 113: 175–182. [ Links ]
18. JIMENEZ–DEL RIO M, VELEZ–PARDO C. The hydrogen peroxide and its importance in Alzheimers and Parkinsons diseases. Curr Med Chem–CNS Agents 2004; 4: 279–285. [ Links ]
19. KERR JFR, WYLLIE AH, CURRIE AR. Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer 1972; 26: 239–257. [ Links ]
20. KERR JFR, GOBE GC, WINTERFORD CM, HARMON BV. Anatomical methods in cell death. En: SCHWARTZ LM, OSBORNE BA, eds. Methods in Cell biology: Cell Death. New York: Academic Press; 1995: 1–27. [ Links ]
21. LEBLANC AC. The role of apoptotic pathways in Alzheimers disease neurodegeneration and cell death. Curr Alzheimer Research 2005; 2: 389–402. [ Links ]
22. MATTSON MP. Apoptosis in neurodegenerative disorders. Nature Rev Mol Cell Biol 2000; 1: 120–129. [ Links ]
23. CHEN N, HUDSON JE, WALCZAK P, MISIUTA I, GARBUZOVA–DAVIS S, JIANG L, et al. Human umbilical cord blood progenitors: The potential of these hematopoietic cells to become neural. Stem Cells 2005; 23: 1.560–1.570. [ Links ]
24. SANCHEZ–RAMOS JR. Neural cells derived from adult bone marrow and umbilical cord blood. J Neurosci Res 2002; 69: 880–893. [ Links ]
25. MARINO F, COSENTINO M, BOMBELLI R, FERRARI M, LECCHINI S, FRIGO G. Endogenous catecholamine synthesis, metabolism, storage, and uptake in human peripheral blood mononuclear cells. Exp Hematol 1999; 27: 489–495. [ Links ]
26. AMENTA F, BRONZETTI E, FELICI L, RICCI A, TAYEBATI SK. Dopamine D2–like receptors on human peripheral blood lymphocytes: a radioligand binding assay and immunocytochemical study. J Auton Pharmacol 1999; 19:151–159. [ Links ]
27. RICCI A, AMENTA F. Dopamine D5 receptors in human peripheral blood lymphocytes: a radioligand binding study. J Neuroimmunol 1994; 53: 1–7. [ Links ]
28. RICCI A, VEGLIO F, AMENTA F. Radioligand binding characterization of putative dopamine D3 receptor in human peripheral blood lymphocytes with {3H} 7OH–DPAT. J Neuroimmunol 1995; 58: 139–144. [ Links ]
29. RICCI A, BRONZETTI E, FELICI L, TAYEBATI SK, AMENTA F. Dopamine D4 receptor in human peripheral blood lymphocytes: a radioligand binding assay study. Neurosci Lett 1997; 229: 130–134. [ Links ]
30. GORDON J, BARNES NM. Lymphocytes transport serotonin and dopamine: agony or ecstasy. Trends Immunol 2003; 24: 438–443. [ Links ]
31. AUNE TM, MCGRATH KM, SARR T, BOMBARA MP, KELLEY KA. Expression of 5HT1a receptors on activated human T cells. Regulation of cyclic AMP levels and T cell proliferation by 5–hydroxytryptamine. J Immunol 1993; 151: 1.175–1.183. [ Links ]
32. CHENNAOUI M, DROGOU C, GOMEZ–MERINO D, GUEZENNEC CY. Effects of an intense training on functional activity of 5–HT(1B) receptors in human peripheral blood lymphocytes. Neurosci Lett 2005; 382: 1–4. [ Links ]
33. FIEBICH BL, AKUNDI RS, SEIDEL M, GEYER V, HAUS U, MULLER W, et al. Expression of 5–HT3A receptors in cells of the immune system. Scand J Rheumatol Suppl 2004; 119: 9–11. [ Links ]
34. KAWASHIMA K, FUJJI T. Extraneural cholinergic system in lymphocytes. Pharmacol Ther 2000; 86: 29–48. [ Links ]
35. LOMBARDI G, DIANZANI C, MIGLIO G, CANONICO PL, FANTOZZI R. Characterization of ionotropic glutamate receptors in human lymphocytes. Br J Pharmacol 2001;133: 936–944. [ Links ]
36. MIGLIO G, VARSALDI F, DIANZANI C, FANTOZZI R, LOMBARDI G. Stimulation of group I metabotropic glutamate receptors evokes calcium signals and cjun and c–fos gene expression in human T cells. Biochem Pharmacol 2005; 70:189–199. [ Links ]
37. SANDERS VM. The role of norepinephrine and beta–2 adrenergic receptor stimulation in the modulation of Th1, Th2, and B lymphocyte function. Adv Exp Med Biol 1998; 437: 269–278. [ Links ]
38. TILLAKARATNE NJ, MEDINA–KAUWE L, GIBSON KM. Gamma aminobutyric acid (GABA) metabolism in mammalian neural and non–neural tissues. Comp Biochem Physiol A Mol Integr Physiol 1995; 112: 247–263. [ Links ]
39. GOMES B, SAVIGNAC M, MOREAU M, LECLERC C, LORY P, GUERY JC, et al. Lymphocyte calcium signaling involves dihydropyridine–sensitive L–type calcium channels: facts and controversies. Crit Rev Immunol 2004; 24: 425–447. [ Links ]
40. KRASZNAI Z. Ion channels in T cells: from molecular pharmacology to therapy. Arch Immunol Ther Exp 2005; 53: 127–135. [ Links ]
41. LUM JB, INFANTE AJ, MAKKER DM, YANG F, BOWMAN BH. Transferrin synthesis by inducer T lymphocytes. J Clin Invest 1986; 77: 841–849. [ Links ]
42. ZAREBSKA Z, ZIELINSKA J, ZHUKOV I, MASLINSKI W. Apoptosis induced by membrane damage in human lymphocytes; effects of arachidonic acid and its photoproducts. Acta Biochem Pol 2005; 52: 179–194. [ Links ]
43. TAPSON VF, SCHENETZLER B, PILCH PF, CENTER DM, BERMAN JS. Structural and functional characterization of the human T lymphocyte receptor for insulin–like growth factor I in vitro. J Clin Invest 1988; 82: 950–957. [ Links ]
44. KOOIJMAN R, WILLEMS M, DECARLA HJC, RIJKERS GT, SCHUURMANS ALG, VAN BUUL–OFFERS SC, et al. Expression of type I insulin–like growth factor receptors on human peripheral blood mononuclear cells. Endocrinology 1992; 131: 2.244–2.250. [ Links ]
45. KANE LP, SHAPIRO VS, STOKOE D, WEISS A. Induction of NFκB by the Akt/ PκB kinase. Curr Biol 1999; 9: 601–604. [ Links ]
46. BIDLACK JM. Detection and function of opioid receptors on cells from the immune system. Clin Diagn Lab Immunol 2000; 7: 719–723. [ Links ]
47. MUNRO S, THOMAS K L, ABU–SHAAR M. Molecular characterization of a peripheral receptor for cannabinoids. Nature (London) 1993; 365: 61–65. [ Links ]
48. LI QX, FULLER SJ, BEYREUTHER K, MASTERS CL. The amyloid precursor protein of Alzheimer disease in human brain and blood. J Leukoc Biol 1999; 66: 567–574. [ Links ]
49. TAKAHASHI N, KARIYA S, HIRANO M, UENO S. Two novel spliced presenilin 2 transcripts in human lymphocyte with oxidant stress and brain. Mol Cell Biochem 2003; 252: 279–283. [ Links ]
50. SUNADA Y, SAITO F, MATSUMURA K, SHIMIZU T. Differential expression of the parkin gene in the human brain and peripheral leukocytes. Neurosci Lett 1998; 254: 180–182. [ Links ]
51. WALKINSHAW G, WATERS CM. Neurotoxin–induced cell death in neuronal PC12 cells is mediated by induction of apoptosis. Neuroscience 1994; 63: 975–987. [ Links ]
52. JIMENEZ DEL RIO M, VELEZ–PARDO C. Monoamine neurotoxin–induced apoptosis in lymphocytes by a common mechanism: involvement of hydrogen peroxide (H2O2), caspase–3, and nuclear factor kappa–B (NF–κB), p53, c–Jun transcription factor. Biochem Pharmacol 2002; 63: 677–688. [ Links ]
53. BEHL C, DAVIS JB, LESLEY R, SCHUBERT D. Hydrogen peroxide mediates amyloid b–protein toxicity. Cell 1994; 77: 817–827. [ Links ]
54. VELEZ–PARDO C, GARCIA OSPINA G, JIMENEZ DEL RIO M. Ab[ 25–35 ] peptide and iron promote apoptosis in lymphocytes by a common oxidative mechanism: involvement of hydrogen peroxide (H2O2), caspase–3, NF–kappa B, p53 and c–Jun. Neurotoxicology 2002; 23: 351–365. [ Links ]
55. MCLELLAN ME, KAJDASZ ST, HYMAN BT, BACSKAI BJ. In vivo imaging of reactive oxygen species specifically associated with thioflavine–S positive amyloid plaques by multiphoton microscopy. J Neurosci 2003; 23: 2.212–2.217. [ Links ]
56. GARCIA–OSPINA G, JIMENEZ DEL RIO M, LOPERA F, VELEZ–PARDO C. El daño nuclear neuronal se correlaciona con la detección in situ de los factores de trascripción c–Jun, factor nuclear–kappa B (FNκB), p53, y Par–4 en
57. JIMENEZ DEL RIO M, MORENO S, GARCIA–OSPINA G, BURITICA O, URIBE CS, LOPERA F, VELEZ–PARDO C. Autosomal recessive juvenile parkinsonism Cys212Tyr mutation in parkin renders lymphocytes susceptible to dopamine and iron–mediated apoptosis. Mov Disorders 2004;19: 324–330. [ Links ]
58. JIMENEZ DEL RIO M, VELEZ–PARDO C. Transition metals–induced apoptosis in lymphocytes via hydroxyl radical generation, mitochondria dysfunction and caspase–3 activation: an in vitro model for neurodegeneration. Arch Med Res 2004; 35:185–193. [ Links ]
59. JIMENEZ DEL RIO C, VELEZ–PARDO C. 17b–estradiol protects lymphocytes against dopamine and ironinduced apoptosis by a genomic–independent mechanism. Implication in Parkinsons disease. Gen Pharmacol 2001; 35: 1–9. [ Links ]
60. JIMENEZ DEL RIO M, VELEZ–PARDO C. Insulin–like growth factor–1 prevents Ab[ 25–35 ] / (H2O2)–induced apoptosis in lymphocytes by reciprocal NF–κB activation and p53 inhibition via PI3K–dependent pathway. Growth Factors 2006; 24: 67–78. [ Links ]
61. VELEZ–PARDO C, JIMENEZ DEL RIO M. Avoidance of Ab[ 25–35 ]???/ (H2O2) –induced apoptosis in lymphocytes by the cannabinoid agonists CP55, 940 and JWH–015 via receptor–independent and PI3Kdependent mechanisms: Role of NF–κB and p53. (Medicinal Chemistry 2006). [ Links ]
Recibido: noviembre 28 de 2005
Aceptado: febrero13 de 2006

