










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.21 no.3 Medellín July/Sept. 2008
ARTÍCULO DE REVISIÓN
Un viejo nuevo gas: el mónoxido de carbono (CO): aspectos esenciales en Biología, Patobiología, Bioclínica y Fármaco–Terapeútica Humana
An old new gas, carbon monoxide (CO): essentials in human Biology, Pathobiology, Bioclinic and human Pharmacotherapeutics
Grégory Alfonso García Morán, MD, candidato a la maestría1; Ananías García Cardona2
1.Docente de la Unidad de Educación y del Instituto de Investigación, Facultad de Medicina, Fundación Universitaria Unisánitas (FUS). Docente del posgrado en Inmunología, Facultad de Ciencias, Pontificia Universidad Javeriana (PUJ), Bogotá, Colombia.
2.Docente y Coordinador de la Unidad de Morfología, Facultades de Medicina y de Rehabilitación, Terapia y Desarrollo Humano y del Instituto de Ciencias Básicas, Universidad Colegio Mayor de Nuestra Señora del Rosario, Bogotá, Colombia.
Correspondencia: Unidad de Educación, Fundación Universitaria Unisánitas (FUS). Avenida (carrera) 68 n.º 22A–30, Ciudadela Sanitaria Sánitas, Bogotá, Colombia. Teléfono 2221500 ext 101.
Direcciones electrónicas: ikarosgreg@gmail.com, gagarcia@unisanitas.edu.co
Resumen
Ha habido grandes avances en el conocimiento de la producción endógena y de las funciones fisiológicas del monóxido de carbono (CO). La mayor parte del CO endógeno se produce en una reacción que puede ser catalizada por tres enzimas denominadas HEMOoxigenasas (HO). La distribución tisular específica de las isoformas de HO (HO–1, HO–2 y HO–3) está muy relacionada con las acciones biológicas del CO como molécula de señalamiento en diferentes sistemas, a saber: neural, vascular (propiedades vasorrelajantes y cardioprotectoras), inmunológico, respiratorio, reproductivo, gastrointestinal, renal y hepático.
El entendimiento de los mecanismos moleculares, celulares, tisulares y sistémicos que regulan la producción y median las acciones fisiológicas del CO provee información sobre los mecanismos patogénicos de muchas enfermedades y estrategias innovadoras para prevenirlas y tratarlas.
Palabras clave:HEMO–oxigenasas (HO), Monóxido de carbono (CO)
Summary
Knowledge of the endogenous production and physiological functions of carbon monoxide (CO) has greatly advanced. Most of the endogenous CO is produced in reactions catalyzed by three enzymes named HEMO–oxygenases (HO). The tissue typespecific distribution of these HO isoforms (HO–1, HO–2 y HO–3) is largely linked to the specific biological actions of CO as a signaling molecule in different systems, namely: neural, vascular (vasorelaxant property and cardiac protection), immunological, respiratory, reproductive, gastrointestinal, kidney, and liver. Understanding the molecular, cellular, tisular and systemic mechanisms that regulate the production and mediate the physiological actions of CO may provide an insight into the pathogenic mechanisms of many diseases, and innovative preventive and therapeutic strategies.
Key words: Carbon monoxide (CO), HEMO–oxigenasas (HO)
INTRODUCCIÓN
La comunicación en los organismos multicelulares es un fenómeno que garantiza la homeostasis, que es el mecanismo base para asegurar la aclimatación como un cambio orquestado, colectivo y cooperativo frente a una variación de las condiciones interiores o exteriores. En 1980 se inició la era del conocimiento de los gases en la trama de la citocomunicación, con la entrada en escena del óxido nítrico (NO), merecedor de una mención 'honorífica' como la molécula del año en 1992.1–4 Se podría denominar técnicamente este tipo de comunicación como 'gasocrina o eolocrina por gasotransmisores'. Posteriormente aparecieron informes sobre el papel del monóxido de carbono (CO) de producción endógena en la trama de la comunicación, a partir del catabolismo del grupo prostético HEMO (también denominado HEME o simplemente HEM), metabolismo especial descrito desde 1968.5,6 Desde 1991 se sugirió una actividad fisiológica para el CO, gracias al estímulo investigativo producido por el NO y los agentes denominados 'relajadores o relajantes endoteliales', pero solo en el año 2000 se comenzó a dilucidar su mecanismo de acción.7,8
Ya se han determinado funciones fisiológicas de otras moléculas gaseosas como el ácido sulfhídrico (H2S) que se deriva del metabolismo del aminoácido L–cisteína,9 el probable rol del metano producido por el metabolismo de las bacterias comensales del intestino,10 el amoníaco (amonio cuando se protona la molécula) derivado del metabolismo aminoacídico11 e incluso el dióxido de carbono (CO2).12
En este manuscrito se revisan sucintamente diversos aspectos del CO y de sus enzimas sintetizadoras –las HEMO–oxigenasas (HO)–, desde la perspectiva de las ciencias básicas y su aplicación a las ciencias clínicas, la farmacología y la terapeútica, con énfasis en la fisiopatología.
METODOLOGÍA
Nuestra búsqueda se sustentó en dos tipos de bancos: los de Genética, Genómica, Proteómica y Enzimología; y los de bibliografía científica. En el primer caso se consultaron el Banco de Genética y Genómica Humana MIM (Mendelian Inheritance McKusick)13 y el HUGO (Human Genome Organization).14 Se utilizarán para los genes, proteínas y trastornos relacionados la nomenclatura y la codificación asignadas por dichos bancos. Para la búsqueda de bibliografía y literatura científica médica humana, se consultaron los dos principales bancos electrónicos: el estadounidense PUBMEDLINE (National Library of Medicine Database)15 y el europeo EMBASE (The Bibliographic Database for Biomedical and Pharmacological Information).16 La matriz de búsqueda que se aplicó para PubMed y EMBASE fue 'Human carbón monoxide (Physiology, Biochemistry, Molecular Cell Biology, Enzimology, Biology, Pathobiology, Immunology, Pathology, Neurobiology, Pharmacology) review', con conector 'or/and', con el límite de fecha '2002', aunque también se tuvieron en cuenta algunas referencias históricas clave.
HISTORIA Y TOXICOLOGÍA DEL CO
La historia del CO necesariamente tiene que ver con su reconocimiento como un tóxico presente en la polución ambiental, que era considerado relativamente insignificante hasta el auge de la industrialización y el urbanismo. Se piensa que el CO es uno de los cinco principales agentes de polución, junto con los sulfóxidos, los compuestos orgánicos volátiles, la materia particulada y los nitróxidos; estos cinco constituyen el 98% del total de los aeroagentes de polución, y el CO es el principal con el 52%, aproximadamente. Probablemente el CO es uno de los principales productos químicos de la polución, puesto que se produce fundamentalmente por reacciones oxidativas en procesos de combustión de materiales orgánicos (sustancias hidrocarbonadas), que son de gran importancia en el mundo industrializado. La concentración atmosférica promedio de CO es de 0,1 partes por millón (ppm), de la cual solo el 10% es producto humano; el 90% restante proviene de múltiples fuentes naturales como la fotooxidación atmosférica del metano, los incendios forestales, las plantas y los microorganismos oceánicos. La principal fuente humana de CO son los automóviles: se calcula que la concentración atmosférica de este gas es de 23 ppm en las áreas residenciales pero alcanza las 115 ppm en lugares con tráfico pesado. Otra fuente humana de CO es el humo del cigarrillo con sus efectos manifiestos sobre la economía y la salud.
Por su masa y por el peso de las partículas, el CO se mantiene principalmente en la baja atmósfera. Los eventos tóxicos son accidentales o suicidas, en sitios con baja ventilación, y se lo ha llamado 'el asesino silente'.
Grandes científicos, como el fisiólogo francés Claude Bernard en 1857 y John Burdon Sanderson Haldane en 1895, describieron la unión del CO a la hemoglobina. La afinidad de este compuesto por la hemoglobina es 210–250 veces más fuerte que la del oxígeno.17
Clásicamente la fisiopatología de la intoxicación por CO es hipóxica, por la formación de carboxihemoglobina, junto con una constricción refleja de la vasculatura pulmonar que impide la oxigenación de los eritrocitos; obviamente, los órganos más vulnerables son los que tienen mayor dependencia del oxígeno, tales como el sistema nervioso central y el corazón, en los que se encuentran tardíamente lesiones de necrosis hemorrágica.
Saint–Martin y Nicloux en 1898 fueron los primeros en describir la producción endógena de CO, y Sjöstrand la demostró en 1951.18,19 Desde 1982 había evidencia de que el CO podía desempeñar roles adicionales en la fisiopatología de las intoxicaciones, en parte porque se unía a los grupos HEMO de proteínas distintas a la hemoglobina, tales como los citocromos y la mioglobina, pero solo en 2000 se dilucidó plenamente ese aspecto.7,8,20
QUÍMICA BIOLÓGICA
El CO es el óxido diatómico del carbono. Es un gas incoloro e inodoro a temperaturas por encima de – 90ºC. Su gravedad específica relativa al aire es 0,967 y su densidad es de 1,25 g/L a la temperatura y presión estándares. Por su triple enlace covalente es una molécula químicamente estable. En condiciones estándar su solubilidad en agua es baja (354 mg/dL). Su capacidad de reaccionar con agua u oxígeno necesita alta energía de activación. Característicamente se produce como parte de una combustión incompleta de la materia orgánica, puesto que si fuera completa se producirían CO2 y agua. La tasa de producción de CO en un ser humano normal es en promedio de 16,4 µmol/h con un máximo de 500 µmol/h.21
BIOSÍNTESIS ENDÓGENA DEL CO: LAS HO
Además de una ruta principal hay varias alternativas para la biosíntesis del CO (Figura n.º 1).
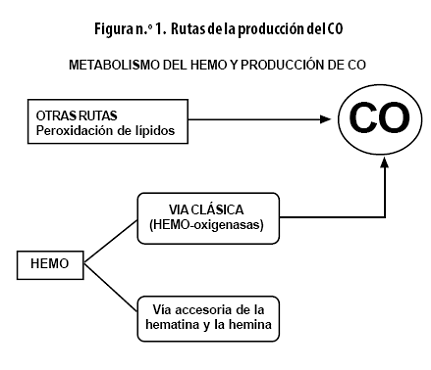
Ruta catabolizadora del HEMO
El catabolismo del HEMO se efectúa oxidativamente por 2 vías:
• Vía clásica dependiente de las HO. Estas son las enzimas limitantes de la tasa en la conversión del HEMO hacia pigmentos biliares.
• Vía accesoria de la hematina y la hemina.
Via clásica dependiente de las HO
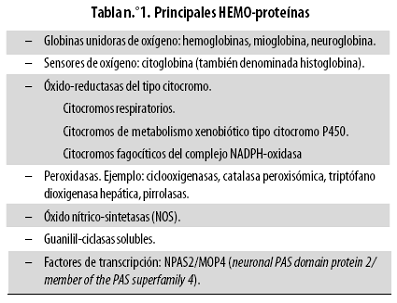
La Unión Internacional de Bioquímica y Biología Molecular (IUBMB, por su sigla inglesa) clasifica la actividad de las HO como EC1.14.99.3 (enzima del grupo 1 [EC1] que corresponde a las óxido–reductasas). Las HO son enzimas esenciales para el metabolismo del grupo HEMO (también denominado técnicamente Ferro–Protoporfirina IX), que funciona como grupo prostético de diversas proteínas –entre otras muchas enzimas de naturaleza óxidoreductasa–, es decir, 'HEMO–proteínas' (Tabla n.º 1).
El grupo HEMO como estructura de la vida está relacionado con fenómenos como el transporte y almacenamiento de oxígeno, el proceso respiratorio mitocondrial productor de energía en forma de adenosín–trifosfato (ATP) y diversos procesos biosintéticos. En otras palabras, sin el grupo HEMO no puede existir la vida.
La reacción catalítica clásica de las HO produce:
• Biliverdina.
• CO.
• Hierro libre ferroso.
• Agua metabólica.
• Coenzima NADP oxidada (Figura n.º 2).

Las HO clivan oxidativamente el HEMO hacia el pigmento biliverdina (biliverdina IXα), el cual es subsecuentemente convertido a bilirrubina (bilirrubina IXα) por intermedio de la enzima biliverdina–reductasa. Este proceso libera colateralmente hierro ferroso y CO. Una de las situaciones más interesantes en la enzimología es el hecho de que el grupo HEMO que se degrada, sea a la vez cofactor catalítico y sustrato de la misma reacción.
Las HO están acopladas a una citocromo P450 reductasa del tipo flavoproteína dependiente de la coenzima NADPH; esto permite entender que el proceso catabólico del HEMO dependa de oxígeno molecular (3 mol de oxígeno por 1 mol de HEMO) y de coenzimas como la nicotinamida. Se ha encontrado in vivo que al complejo HO–citocromo reductasa está acoplada la biliverdina–reductasa, que es una enzima citosólica.
Las HO comparten una homología proteica del 80%. Dependiendo de sus patrones de expresión se pueden clasificar grosso modo como:
• Constitutivas: HO–2 y HO–3.
• Inducible: HO–1.
La variante inducible HO–1 se denomina así porque diversas respuestas de estrés estimulan su expresión; sin embargo, HO–1 también es constitutiva en el retículo–endotelio ( hígado, bazo, médula ósea y mesangio renal). La variante constitutiva de expresión no inmune es la HO–2, que prácticamente se expresa en todos los tipos celulares. Una tercera variante es la HO–3, inicialmente descubierta como una chaperonina, es decir, una proteína pequeña moldeante; más tarde se determinó su relación con las HO y se le encontró una expresión constitutiva. La HO–3 tiene una baja actividad enzimática lo cual hace pensar que sea un barredor o sensor del HEMO. Hay aún algunas incertidumbres sobre la bioquímica y la existencia de HO–3. En las tablas n.º 2 y 3 se consignan datos referentes a la genética, genómica, patrones de expresión y patobiología de las HO.
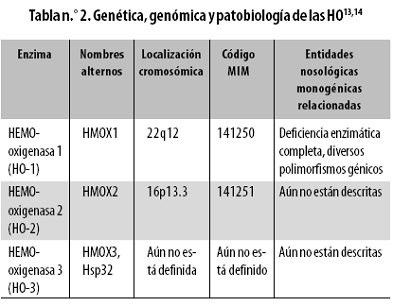
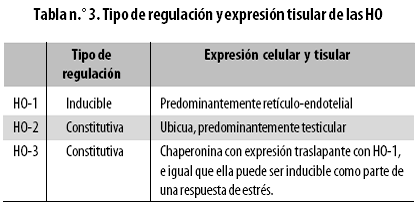
Las HO son enzimas intracelulares con patrones complejos de expresión; tradicionalmente siempre se ha considerado que la HO–1 es una enzima soluble del retículo endoplásmico liso (o microsomal) y la HO–2, una enzima anclada a la membrana del mismo organelo; pero hoy es evidente que la HO–1 se encuentra o es movilizada activamente hacia casi cualquier compartimento intracelular, y que la HO–2 puede incluso ser una proteína de la membrana celular plasmática (plasmalema).22,23
Filogenéticamente, las HO son enzimas muy difundidas en todos los reinos vivientes, e incluso se encuentran en ciertas bacterias en cuya adquisición de hierro exógeno desempeñan roles importantes. Sin embargo, hoy también se sustenta el papel patógeno de ciertas bacterias, protozoarios y hongos, mediante sus propias HO.24
Vía accesoria de la hematina y la hemina
Las rutas de la hematina y la hemina no están totalmente entendidas en el contexto fisiológico y bioquímico humano; parecería que fueran fundamentalmente anenzimáticas (sin necesidad de enzimas), pero es importante mencionar que la hemina por sí sola ha mostrado ser un potente eritropoyético e inductor globínico. Clásicamente se han encontrado estos compuestos como inclusiones intracelulares, fruto de la infección con protozoarios, pero se los halla también en el metabolismo alterno normal del HEMO.25
Otras rutas
Además del metabolismo del HEMO, hay informes de otras fuentes productoras de CO, a saber: la autooxidación y la oxidación enzimática de fenoles, la fotooxidación de compuestos orgánicos y la peroxidación de lípidos. Por otra parte, el fenobarbital y la difenilhidantoína aumentan la producción de este compuesto. Así mismo, se puede formar CO cuando se usan agentes anestésicos volátiles como el desfluorano y el sevofluorano con sistemas de respiración que contienen absorbentes para CO2.26
MECANISMOS DE REGULACIÓN DE LAS HO
La regulación de la expresión génica y de la actividad de las HO es compleja y ha sido difícil integrar en un solo modelo toda la información disponible, puesto que cada célula y cada tejido tienen distintos mecanismos de regulación que pueden variar en el tiempo. En el caso particular de la enzima inducible, es larga la lista de inductores y están involucradas prácticamente todas las rutas de señalamiento intracelular de transducción; se puede decir que cualquier factor es capaz de desencadenar una respuesta de estrés con activación final de la HO–1. Por otra parte, para la HO–1 son inductores fisiológicos el HEMO y sus derivados, la hipoglicemia y el CO per se (Tabla n.º 4).

Si bien la HO–2 es constitutiva, no escapa a cierta regulación positiva por parte de los glucocorticoides, los opiáceos, el glutamato y los estrógenos, y recibe un influjo negativo mediado por el NO y el CO, también per se. Las rutas de señalamiento reguladoras de HO–2 involucran el calcio, las proteínas–quinasas C (PQC) y las calmodulinas; estas últimas se unen directamente a la HO–2 y la activan.27,28
REGULACIÓN GÉNICA DE LA HO–1: UN SISTEMA HEMODEPENDIENTE
En las células de los mamíferos se produce el fascinante fenómeno de que el HEMO, sus derivados porfirínicos y el CO regulan a varios niveles la expresión génica de diversas enzimas, su anabolismo, su catabolismo y su actividad. Así, por ejemplo, el HEMO funciona como un segundo mensajero intracelular que se une a unas proteínas muy especiales denominadas Bach (basic leucine zipper transcription factor/ BTB and CNC homology), de las cuales se conocen, en el Homo sapiens, Bach1 y Bach2. Las proteínas Bach son factores de transcripción que entran al núcleo y que, tras asociarse con otras proteínas intranucleares, se unen en conjunto al promotor de diversos genes y reprimen su expresión, es decir, su transcripción hacia un ARN mensajero que pudiera ser traducido ulteriormente hacia una proteína. El gen reprimido por excelencia es el que codifica HO–1, pero también lo son otros como el de la betaglobina.
Las proteínas de asociación pertenecen a una familia de protooncogenes inicialmente descritos en especies aviares, es decir, genes promotores de la oncogénesis o neoplasiogénesis, y se denominan MAF (v–MAF musculoaponeurotic fibrosarcoma oncogene homologous). En el Homo sapiens se ha definido que existen seis miembros cuya expresión varía dependiendo, entre otros factores, de los tipos celulares, el grado de diferenciación y el tipo de tejido. El mecanismo es el siguiente:
•1. Los complejos Bach/MAF reprimen la expresión del gen codificante de HO–1.
•2. El HEMO disponible entra al núcleo y se une a las proteínas Bach, haciéndoles cesar su actividad represora.
•3. Se estimula la expresión de genes involucrados en la respuesta celular y tisular a CO puesto que las MAF se asocian ahora con factores estimuladores de la transcripción denominados NFE (nuclear factor erythroid/ NFE2–related factor), de los cuales se conocen tres en los seres humanos: NRF1, NRF2 y NRF3), y se recluta una subunidad accesoria adicional denominada p45NFE2. Para esto los NRF, que se ubican en el citosol junto con la proteína inhibidora KEAP1 (kelch–like ECH–associated protein 1), tras la activación de toda la ruta, se desacoplan del inhibidor proteico y son movilizados hacia el núcleo (Figura n.º 3). Todo esto ocurre porque la expresión reducida de HO–1 puede ayudar a preservar el HEMO intracelular como un componente importante para ensamblar otras HEMOproteínas y, de paso, reducir el gasto energético oxidativo catabólico.29,30

MECANISMOS BÁSICOS DE LA DINÁMICA DE ACCIÓN DEL CO
El CO es una molécula altamente estable y no reactiva, a diferencia de su congénere el NO, que es un radical libre. La afinidad y especificidad de unión del CO por las HEMO–proteínas son altas, uniéndose a núcleos ferro–HEMO (es decir, con un átomo de hierro reducido) a diferencia del NO que se une a núcleos ferri–HEMO (es decir, con un átomo de hierro oxidado). Esto hace que pueda interactuar con las hemoproteínas previamente mencionadas (sean o no enzimas) y regular o modificar su actividad total. En la tabla n.º 5 se resumen los efectos activador e inhibidor del CO sobre diversas HEMO–proteínas.
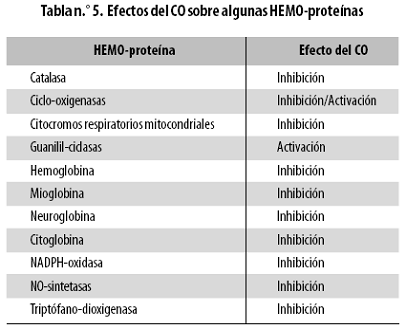
El CO está constantemente siendo oxidado, barrido y espirado en nuestro organismo, de tal manera que se podría decir que hay una metaestabilidad constante entre lo producido y lo catabolizado.28
ROLES BIOLÓGICOS Y PATOBIOLÓGICOS DEL CO
La actividad del CO se fundamenta en cuatro hechos:
• 1. Actúa como primer y segundo mensajeros en la comunicación celular.28,31
• 2. En muchos estudios se ha demostrado que la inducción de HO–1 y la producción de CO son antiinflamatorias, antiapoptóticas, antiproliferativas y citoprotectoras. Es claro que la inducción de HO–1 en casi cualquier órgano forma parte de la llamada 'respuesta homeostática de estrés'. Muchos de estos efectos se basan en la inhibición de HEMO–proteínas como las ciclooxigenasas, la NADPH–oxidasa y las NO–sintetasas, enzimas que producen mediadores proinflamatorios y que colateralmente desencadenan fenómenos oxidativos y proapoptóticos.28,32–34
• 3. La activación de las guanilil–ciclasas solubles aumenta la biosíntesis de guanosina–monofosfato cíclico (GMPc) que a su vez se une y activa la enzima fosforilante PQG (proteína quinasa G). La PQG fosforila diversos sustratos que ejercen un efecto tampón frente al calcio. El GMPc también modula directamente la actividad de otras enzimas (por ejemplo, nucleótido cíclico fosfodiesterasas) y canales iónicos.28,35
• 4. La interacción directa del CO con algunos canales iónicos, y en especial con los de alta conductancia para potasio llamados BKCa (Big conductance K Ca channels). La activación de estos canales se debe a una interacción específica de la subunidad α con el CO, a diferencia de la subunidad β que es reconocida por el NO, al cual se une. Además, el grupo HEMO y la HO–2 per se pueden unirse directamente a estos canales y regular su función.28,36,37
• Los cuatro hechos anteriores son la base de varias funciones fisiológicas del sistema HO–CO, tales como (Tabla nº 6):
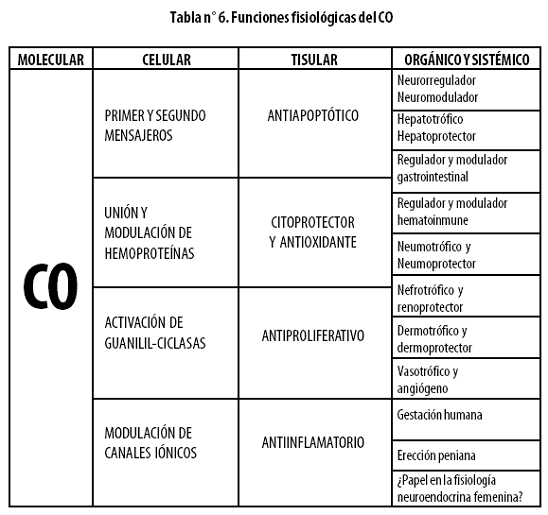
– Citoprotector general prorregenerativo, prodiferenciador y trófico madurador.38
– Es un neurotransmisor implicado en la potenciación sináptica a largo plazo (LTP, long term potentiation) y, por ende, en la memoria y el aprendizaje, la función neurorretiniana, la relajación muscular no adrenérgica y no colinérgica, la regulación colinérgica de los ritmos circadianos, la regulación de la temperatura, la regulación autonómica de la función cardiovascular y la respuesta adaptadora olfatoria (olores) y vomeronasal (feromonas odoríferas). Está relacionado con los procesos de nocicepción y quimiorrecepción (procesos de detección de oxígeno en el tallo cerebral). Es también un regulador positivo de la liberación de neuropéptidos en el sistema hipotálamo–hipofisiario, en particular un modulador del eje hipotálamo–hipófisis–corteza suprarrenal.39–44
– Es un hepatotrófico y hepatoprotector: HO–1 es inducida por procesos de isquemia/reperfusión, metales pesados, etanol, radicales libres, acetaminofén, choque hemorrágico, hipoxia, endotoxinas y algunos fármacos anestésicos (halotano e isofluorano).45–47
– Desempeña papeles importantes en la fisiología gastrointestinal: modula el recambio epitelial enterocítico, la diferenciación celular, la detoxificación y la inflamación. Es un supresor de la oncogénesis y regula los procesos de absorción y motilidad. Muestra actividad protectora del páncreas.48,49
– Desde el punto de vista de la hematología y la inmunidad es un antiinflamatorio en diversos sitios y un antiagregante plaquetario; desempeña roles importantes en el metabolismo del hierro y es un inmunomodulador. Con respecto a esta última actividad, es evidente que inhibe tanto respuestas inmunes de tipo celular (del brazo linfocitario ayudador 1 –Th1–) como de tipo humoral (del brazo linfocitario ayudador 2 –Th2–) y propende por la activación de la tercera vía, es decir, la de tolerancia mediante las células T reguladoras (TREG).34,50–52
– Es neumotrófico y protector pulmonar,53–56 nefrotrófico y protector renal,57–60 dermotrófico y protector de la piel.61
– Tiene efectos vasoactivos y vasotróficos como vasodilatador, proangiogénico y antiarterioesclerosis. El factor de crecimiento vascular endotelial A (VEGFA) es un inductor de HO–1.62–64
– Durante la gestación, por ser tocolítico, está implicado en el mantemiento del útero quiescente, en el control hemodinámico, en la regulación de las cascadas apoptóticas e inflamatorias del trofoblasto, y en el mantenimiento del equilibrio oxidante/antioxidante.65
– El CO, al igual que su familiar el NO, regula la erección peniana, pero con un papel secundario en tal evento biológico. 66
– Hay evidencia indirecta, a partir de murinos, del papel del sistema HO/CO en la neuroendocrinología en todos los niveles del eje gonadotropo femenino, desde el hipotálamo hasta los órganos sexuales.67
PATOBIOLOGÍA Y BIOCLÍNICA DEL SISTEMA HO/CO
A la luz de lo anterior se puede concebir que la disregulación del mecanismo homeostático de este sistema subyace a casi todos los trastornos conocidos, desde los neoplásicos, pasando por los infecciosos y llegando a los puramente disfuncionales de índole vascular y neural.68,69 Como si fuera poco, desde 1997 se han identificado variaciones genéticas (polimorfismos génicos) en el gen de HO–1 que causan disfunción de la actividad del gen; en otras palabras, la enzima producida es baja sintetizadora de CO; dichas variaciones se relacionan con una mayor predisposición a enfermedades neoplásicas, vasculares, inflamatorias (infecciosas, autoinmunes) y neurológicas, así como a una evolución más virulenta de las mismas. Otro tipo de polimorfismo génico está vinculado a una mayor producción de CO, lo que podría ser un factor clave en la resistencia a varias enfermedades y la tolerancia postrasplantes. Se han descubierto variaciones génicas protectoras o aceleradoras del envejecimiento en el sistema HO/CO. 70,71
Curiosamente, también se ha identificado la deficiencia total de HO–1. Fue el caso particular de un paciente de 26 meses descrito en 1999, con un cuadro clínico polimorfo caracterizado por un trastorno difuso con manifestaciones de fiebre recurrente y un exantema eritematoso generalizado, retardo del crecimiento, anemia hemolítica, reticuloendoteliosis, hemocromatosis y extrema vulnerabilidad a lesiones de cualquier índole (infecciosas, xenobióticas).72 Es muy llamativa la hemocromatosis, que ha abierto un nuevo campo de investigación con respecto al sistema HO/CO en el metabolismo del hierro.73,74
FARMACOLOGÍA Y TERAPÉUTICA
El sistema descrito ofrece posibilidades terapeúticas reales para una gran diversidad de trastornos en los que aminora las lesiones. También, por favorecer procesos de tolerancia, amplía las perspectivas en la medicina de trasplantes y la implantología.68,69,75
Entonces, lo ideal es regular positivamente el sistema. Las posibilidades que para ello se están investigando son las siguientes:
– Terapia génica: inserción del gen mediante vectores. Lograda desde 1995 en conejos pero aún en experimentación para usos futuros en seres humanos.76
– Terapia no génica.
– Nutriceúticos inductores del sistema HO/CO.77
– Inhalación de CO.78
– Inductores de HO como el NO y sus derivados, al igual que HEMO–derivados.79
– Compuestos liberadores de CO (CO–RM) [del inglés CO–Releasing Materials]: metalcarbonilos en desarrollo con base en manganeso (CORM–1), rutenio (CORM–2 and –3), boro (CORM–A1) y hierro (CORM–F3).80,81
–Prodrogas generadoras de CO: diclorometano (DCM) y afines.82
Bajo ciertas circunstancias se busca lo contrario, o sea, inhibir el sistema en casos de intoxicación férrica y de trastornos genéticos del metabolismo de las porfirinas, es decir, en las porfirias. Tal intoxicación férrica es un factor clave en la patobiología de ciertas enfermedades neurodegenerativas.73,74,83–86
CONCLUSIÓN
Se ha demostrado que el sistema HO/CO es fundamental como proceso homeostásico. Conocerlo permite entender a fondo el comportamiento fisiológico y patológico. Los avances en farmacoterapia del sistema HO/CO abren nuevas opciones para el mejor tratamiento de varias enfermedades.
REFERENCIAS BIBLIOGRÁFICAS
1. Perbal B. Communication is the key. Cell Commun Signal 2003; 1: 3–4. [ Links ]
2. Koshland DE. The molecule of the year. Science 1992; 258: 1861. [ Links ]
3. Culotta E, Koshland DE. NO news is good news. Science 1992; 258: 1862–1865. [ Links ]
4. Ray A, Chakraborti A, Gulati K. Current trends in nitric oxide research. Cell Mol Biol 2007; 53: 3–14. [ Links ]
5. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA 1968; 61: 748–755. [ Links ]
6. Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem 1969; 244: 6388–6394. [ Links ]
7. Marks GS, Brien JF, Nakatsu K, McLaughlin BE. Does carbon monoxide have a physiological function? Trends Pharmacol Sci 1991; 12: 185–188. [ Links ]
8. Ortiz de Montellano PR. The mechanism of heme oxygenase. Curr Opin Chem Biol 2000; 4: 221–227. [ Links ]
9. Pearson RJ, Wilson T, Wang R. Endogenous hydrogen sulfide and the cardiovascular system. What's the smell all about? Clin Invest Med 2006; 29: 146–150. [ Links ]
10. Kurbel S, Kurbel B, Vcev A. Intestinal gases and flatulence: possible causes of occurrence. Med Hypotheses 2006; 67: 235–239. [ Links ]
11. Rose C. Effect of ammonia on astrocytic glutamate uptake/release mechanisms. J Neurochem 2006; 97 (Suppl. 1): 11–15. [ Links ]
12. Shimokawa N, Dikic I, Sugama S, Koibuchi N. Molecular responses to acidosis of central chemosensitive neurons in brain. Cell Signal 2005; 17: 799–808. [ Links ]
13. OMIM [base de datos en Internet]. Baltimore: Johns Hopkins University; 1966– [fecha de acceso 15 de julio del 2007]. Disponible en: http://www.ncbi.nlm.nih.gov/entrez/dispomim. [ Links ]
14. HUG[base de datos en Internet]. Bethesda: National Library of Medicine and others (exp.: Celera Genomics and the Sanger Center); 1989– [fecha de acceso 15 de julio del 2007]. Disponible en: http://www.huginternational.org/index.html. [ Links ]
15. PubMed [base de datos en Internet]. Bethesda: National Library of Medicine; 1966– [fecha de acceso 15 de julio del 2007]. Disponible en: http://www.ncbi.nlm.nih.gov/PubMed/. [ Links ]
16. EMBASE [base de datos en internet]. Amsterdam: Elsevier B.V.; 1974– [fecha de acceso 15 de julio del 2007]. Disponible en: http://www.info.embase.com/embase_com/. [ Links ]
17. Gorman D, Drewry A, Huang YL, Sames C. The clinical toxicology of carbon monoxide. Toxicology 2003; 187: 25–38. [ Links ]
18. Sjöstrand T. Endogenous formation of carbon monoxide: the CO concentration in the inspired and expired air of hospital patients. Acta Physiol Scand 1951; 22: 137–141. [ Links ]
19. Sjöstrand T. A preliminary report on the in vitro formation of carbon monoxide in blood. Acta Physiol Scand 1951; 22: 142–143. [ Links ]
20. Gutierrez G. Carbon monoxide toxicity. Air pollutionphysiological effects. McGrath JJ, Barnes CD, eds. New York: Academic Press; 1982, pp. 127–147. [ Links ]
21. Piantadosi CA. Biological chemistry of carbon monoxide. Antioxid Redox Signal 2002; 4: 259–270. [ Links ]
22. Poulos TL. Structural biology of heme monooxygenases. Biochem Biophys Res Commun 2005; 338: 337–345. [ Links ]
23. Maines MD. The heme oxygenase system: update 2005. Antioxid Redox Signal 2005; 7: 1761–1766. [ Links ]
24. Frankenberg–Dinkel N. Bacterial heme oxygenases. Antioxid Redox Signal 2004; 6: 825–834. [ Links ]
25. Tsiftsoglou AS, Tsamadou AI, Papadopoulou LC. Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacol Ther 2006; 111: 327–345. [ Links ]
26. Coppens MJ, Versichelen LF, Rolly G, Mortier EP, Struys MM. The mechanisms of carbon monoxide production by inhalational agents. Anaesthesia 2006; 61: 462–468. [ Links ]
27. Ryter SW, Otterbein LE, Morse D, Choi AM. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem 2002; 1–2: 234–235; 249–263. [ Links ]
28. Ryter SW, Alam J, Choi AM. Heme oxygenase–1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006; 86: 583–650. [ Links ]
29. Jeong WS, Jun M, Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal 2006; 8: 99–106. [ Links ]
30. Igarashi K, Sun J. The heme–Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal 2006; 8: 107–118. [ Links ]
31. Kim HP, Ryter SW, Choi AM. CO as a cellular signaling molecule. Annu Rev Pharmacol Toxicol 2006; 46: 411–449. [ Links ]
32. Wagener FA, Volk HD, Willis D,Soares MP, Adema GJ, Figdor CG. Different faces of the heme–heme oxygenase system in inflammation. Pharmacol Rev 2003; 55: 551–571. [ Links ]
33. Alcaraz MJ, Fernandez P, Guillen MI. Anti–inflammatory actions of the heme oxygenase–1 pathway. Curr Pharm Des 2003; 9: 2541–2551. [ Links ]
34. Mannaioni PF, Vannacci A, Masini E. Carbon monoxide: the bad and the good side of the coin, from neuronal death to anti–inflammatory activity. Inflamm Res 2006; 55: 261–273. [ Links ]
35. Jackson EB Jr, Mukhopadhyay S, Tulis DA. Pharmacologic modulators of soluble guanylate cyclase/cyclic guanosine monophosphate in the vascular system – from bench top to bedside. Curr Vasc Pharmacol 2007; 5: 1–14. [ Links ]
36. Wu SN. Large–conductance Ca2+–activated K+ channels: physiological role and pharmacology. Curr Med Chem 2003; 10: 649–661. [ Links ]
37. Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, et al. Heme is a carbon monoxide receptor for largeconductance Ca2+–activated K+ channels. Circ Res 2005; 97: 805–812. [ Links ]
38. Wagener FA, van Beurden HE, von den Hoff JW, Adema GJ, Figdor CG. The heme–heme oxygenase system: a molecular switch in wound healing. Blood 2003; 102: 521–528. [ Links ]
39. Raub JA, Benignus VA. Carbon monoxide and the nervous system. Neurosci Biobehav Rev 2002; 26: 925–940. [ Links ]
40. Mancuso C, Perluigi M, Cini C, De Marco AM, Giunffrida S, Calabrese V. Heme oxygenase and cyclooxygenase in the central nervous system: a functional interplay. J Neurosci Res 2006; 84: 1385–1391. [ Links ]
41. Mancuso C. Heme oxygenase and its products in the nervous system. Antioxid Redox Signal 2004; 6: 878–887. [ Links ]
42. Schipper HM. Heme oxygenase expression in human central nervous system disorders. Free Radic Biol Med 2004; 37: 1995–2011. [ Links ]
43. Orozco–Ibarra M, Chirino YI, Pedraza–Chaverrí J. Role of hemeoxygenase–1 in the neurodegenerative disorders. Rev Neurol 2006; 43: 556–562. [ Links ]
44. Cutajar MC, Edwards TM. Evidence for the role of endogenous carbon monoxide in memory processing. J Cogn Neurosci 2007; 19: 557–562. [ Links ]
45. Wunder C, Potter RF. The heme oxygenase system: its role in liver inflammation. Curr Drug Targets Cardiovasc Haematol Disord 2003; 3: 199–208. [ Links ]
46. Suematsu M, Tsukada K, Tajima T, Yamamoto T, Ochiai D, Watanabe H, et al. Carbon monoxide as a guardian against hepatobiliary dysfunction. Alcohol Clin Exp Res 2005; 29 (Suppl. 11): S134–S139. [ Links ]
47. Farombi EO, Surh YJ. Heme oxygenase–1 as a potential therapeutic target for hepatoprotection. J Biochem Mol Biol 2006; 39: 479–491. [ Links ]
48. Gibbons SJ, Farrugia G. The role of carbon monoxide in the gastrointestinal tract. J Physiol 2004; 556: 325–336. [ Links ]
49. Oates PS, West AR. Heme in intestinal epithelial cell turnover, differentiation, detoxification, inflammation, carcinogenesis, absorption and motility. World J Gastroenterol 2006; 12: 4281–4295. [ Links ]
50. Gende OA. Carbon monoxide inhibits capacitative calcium entry in human platelets. Thromb Res 2004; 114: 113–119. [ Links ]
51. Brusko TM, Wasserfall CH, Agarwal A. An integral role for heme oxygenase–1 and carbon monoxide in maintaining peripheral tolerance by CD4+CD25+ regulatory T cells. J Immunol 2005; 174: 5181–5186. [ Links ]
52. Bach FH. Heme oxygenase–1 and transplantation tolerance. Hum Immunol 2006; 67: 430–432. [ Links ]
53. Ryter SW, Choi AM. Therapeutic applications of carbon monoxide in lung disease. Curr Opin Pharmacol 2006; 6: 257–262. [ Links ]
54. Sampsonas F, Karkoulias K, Kaparianos A, Spiropoulos K. Genetics of chronic obstructive pulmonary disease, beyond a1–antitrypsin deficiency. Curr Med Chem 2006; 13: 2857–2873. [ Links ]
55. Fredenburgh LE, Perrella MA, Mitsialis SA. The role of heme oxygenase–1 in pulmonary disease. Am J Respir Cell Mol Biol 2007; 36: 158–165. [ Links ]
56. Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol 2007; 36: 175–182. [ Links ]
57. Calo LA, Davis PA, Piccoli A, Pessina AC. A role for heme oxygenase–1 in the antioxidant and antiapoptotic effects of erythropoietin: the start of a good news/bad news story? Nephron Physiol 2006; 103: 107–111. [ Links ]
58. Li Volti G, Rodella LF, Di Giacomo C, Rezzani R, Bianchi R, Borsani E, et al. Role of carbon monoxide and biliverdin in renal ischemia/reperfusion injury. Nephron Exp Nephrol 2006; 104: e135–139. [ Links ]
59. Nath KA. Heme oxygenase–1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 2006; 70: 432–443. [ Links ]
60. Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 2007; 18: 414–420. [ Links ]
61. Wojas–Pelc A, Marcinkiewicz J. What is a role of haeme oxygenase–1 in psoriasis? Current concepts of pathogenesis. Int J Exp Pathol 2007; 88: 95–102. [ Links ]
62. Bussolati B, Mason JC. Dual role of VEGF–induced hemeoxygenase–1 in angiogenesis. Antioxid Redox Signal 2006; 8: 1153–1163. [ Links ]
63. Durante W, Johnson FK, Johnson RA. Role of carbon monoxide in cardiovascular function. J Cell Mol Med 2006; 10: 672–686. [ Links ]
64. Immenschuh S, Schroder H. Heme oxygenase–1 and cardiovascular disease. Histol Histopathol 2006; 21: 679–685. [ Links ]
65. Bainbridge SA, Smith GN. HO in pregnancy. Free Radic Biol Med 2005; 38: 979–988. [ Links ]
66. Watts GF, Chew KK, Stuckey BG. The erectile–endothelial dysfunction nexus: new opportunities for cardiovascular risk prevention. Nat Clin Pract Cardiovasc Med 2007; 4: 263–273. [ Links ]
67. Alexandreanu IC, Lawson DM. Heme oxygenase in the rat anterior pituitary: immunohistochemical localization and possible role in gonadotropin and prolactin secretion. Exp Biol Med 2003; 228: 64–49. [ Links ]
68. Morse D, Sethi J. Carbon monoxide and human disease. Antioxid Redox Signal 2002; 4: 331–338. [ Links ]
69. Ryter SW, Otterbein LE. Carbon monoxide in biology and medicine. Bioessays 2004; 26: 270–280. [ Links ]
70. Kimpara T, Takeda A, Watanabe K, Itoyama Y, Ikawa S, Watanabe M, et al. Microsatellite polymorphism in the human heme oxygenase–1 gene promoter and its application in association studies with Alzheimer and Parkinson disease. Hum Genet 1997; 100: 145–147. [ Links ]
71. Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase–1 promoter polymorphisms in human disease. Free Radic Biol Med 2004; 37: 1097–1104. [ Links ]
72. Koizumi S. Human heme oxygenase–1 deficiency: a lesson on serendipity in the discovery of the novel disease. Pediatr Int 2007; 49: 125–132. [ Links ]
73. Balla J, Vercellotti GM, Jeney V , Akihiro Yachie A, Varga Z, John W. Eaton JW, et al. Heme, heme oxygenase and ferritin in vascular endothelial cell injury. Mol Nutr Food Res 2005; 49: 1030–1043. [ Links ]
74. Lee DW, Andersen JK, Kaur D. Iron dysregulation and neurodegeneration: the molecular connection. Mol Interv 2006; 6: 89–97. [ Links ]
75. Ryter SW, Choi AM. Cytoprotective and anti–inflammatory actions of carbon monoxide in organ injury and sepsis models. Novartis Found Symp 2007; 280: 165–175. [ Links ]
76. Abraham NG, Asija A, Drummond G, Peterson S. Heme oxygenase–1 gene therapy: recent advances and therapeutic applications. Curr Gene Ther 2007; 7: 89–108. [ Links ]
77. Ogborne RM, Rushworth SA, Charalambos CA, O'Connell MA. Haem oxygenase–1: a target for dietary antioxidants. Biochem Soc Trans 2004; 32: 1003–1035. [ Links ]
78. Resch H, Zawinka C, Weigert G, Schmetterer L, Garhöfer G. Inhaled carbon monoxide increases retinal and choroidal blood flow in healthy humans. Invest Ophthalmol Vis Sci 2005; 46: 4275–4280. [ Links ]
79. Schroder H. No nitric oxide for HO–1 from sodium nitroprusside. Mol Pharmacol 2006; 69: 1507–1509. [ Links ]
80. Foresti R, Shurey C, Ansari T, Sibbons P, Mann BE, Johnson TR, et al. Reviewing the use of carbon monoxide–releasing molecules (CO–RMs) in biology: implications in endotoxin–mediated vascular dysfunction. Cell Mol Biol 2005; 51: 409–423. [ Links ]
81. Motterlini R, Mann BE, Foresti R. Therapeutic applications of carbon monoxide–releasing molecules. Expert Opin Investig Drugs 2005; 14: 1305–1318. [ Links ]
82. Martins PN, Reutzel–Selke A, Jurisch A, Denecke C, Attrot K, Pascher A, et al. Induction of carbon monoxide in donor animals prior to organ procurement reduces graft immunogenicity and inhibits chronic allograft dysfunction. Transplantation 2006; 82: 938–944. [ Links ]
83. Vreman HJ, Cipkala DA, Stevenson DK. Characterization of porphyrin heme oxygenase inhibitors. Can J Physiol Pharmacol 1996; 74: 278–285. [ Links ]
84. Grundemar L. Pitfalls using metalloporphyrins in carbon monoxide research. Trends Pharmacol Sci 1997; 18: 193–195. [ Links ]
85. Foresti R, Green CJ, Motterlini R. Generation of bile pigments by haem oxygenase: a refined cellular strategy in response to stressful insults. Biochem Soc Symp 2004; 71: 177–192. [ Links ]
86. Mancuso C, Pani G, Calabrese V. Bilirubin: an endogenous scavenger of nitric oxide and reactive nitrogen species. Redox Rep 2006; 11: 207–213. [ Links ]
Recibido: enero 13 de 2008
Aceptado: marzo 12 de 2008

