










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.21 no.4 Medellín Oct./Dec. 2008
ARTÍCULO DE REVISIÓN
Encefalopatía hepática
Hepatic encephalopathy
Juan Pablo Londoño Múnera1; Lorena Bejarano Pineda1; Juan Carlos Restrepo Gutiérrez2
1 Estudiante de Medicina de la Universidad de Antioquia, Joven investigador del grupo de Gastrohepatología.
2 MD, MSc, PhD. Profesor Asociado, Universidad de Antioquia, Grupo de Gastrohepatología, Unidad de Hepatología Hospital Pablo Tobón Uribe, Medellín, Colombia.
Dirección de contacto: jcrestrepo@hptu.org.co
Resumen
En pacientes con enfermedad hepática aguda o crónica la encefalopatía es una complicación progresiva, aunque la mayoría de las veces reversible, que puede reducir la expectativa de vida y deteriorar su calidad. En cuanto a la patogénesis, se han planteado diferentes hipótesis, la más aceptada de las cuales ha sido la neurotoxicidad por amonio, que incluso podría explicar otras como la del edema de los astrocitos y la toxicidad por manganeso. La clasificación actual de la encefalopatía hepática, propuesta en el Congreso Mundial de Gastroenterología en 1998, se basa en la naturaleza de la disfunción hepática y en la duración y características de las manifestaciones neurológicas. Para su diagnóstico, que es principalmente de exclusión, se requiere un buen conocimiento clínico; sin embargo, actualmente se cuenta con los criterios de West Haven para clasificar su gravedad. El tratamiento de los pacientes con este síndrome, para controlar las manifestaciones neurológicas y mejorar la calidad de vida, se basa en aplicar diferentes medidas como la dieta proteica, los disacáridos no absorbibles y la administración de antibióticos; sin embargo, la principal medida terapéutica es el control de los factores precipitantes de la encefalopatía.
El objetivo de este artículo fue revisar los conceptos recientes sobre la encefalopatía hepática, con especial énfasis en su patogénesis, clasificación, diagnóstico y tratamiento.
Palabras clave: Encefalopatía hepática, Criterios de West Haven, Dieta proteica, Neurotoxicidad por amonio, Rifaximin
Summary
Encephalopathy is a progressive but reversible complication in patients with acute or chronic hepatic disease. It can reduce the expectancy of life and affect its quality. The most accepted pathogenic theory to explain this syndrome is that of the ammonium toxicity, which may also explain other hypotheses such as edema of the astrocytes, and magnesium toxicity. The present classification system for hepatic encephalopathy was proposed in 1998 at the World Congress of Gastroenterology. It is based on the nature of the hepatic dysfunction, and the characteristics of the neurological manifestations. In order to make the diagnosis, a good clinical knowledge is necessary, since it is based mainly on the exclusion of other diseases. The West Haven criteria are presently used to classify the severity of its clinical presentation. To control the manifestations of this syndrome and to improve the quality of life of patients, different therapeutic measures are used, namely: a diet that includes proteins, non–absorbable disaccharides, and the administration of antibiotics; however, the most important therapeutic measure is to control the precipitating factors. The aim of this article was to review recent concepts on hepatic encephalopathy, with emphasis on its pathogenesis, classification, diagnosis, and treatment.
Key words: Ammonium neurotoxicity, Hepatic encephalopathy, Proteic diet, Rifaximin, West Haven criteria
INTRODUCCIÓN
La encefalopatía hepática (EH) es una complicación neuropsiquiátrica grave y progresiva, pero potencialmente reversible, que ocurre en pacientes con falla hepática aguda y en los que tienen enfermedad hepática crónica avanzada. Abarca una serie de manifestaciones que van desde una alteración leve de las funciones motora y cognitiva hasta el deterioro grave del estado de conciencia, el coma y la muerte.1,2
Se ha encontrado en diferentes estudios3,4 que la EH reduce la expectativa de vida de pacientes con enfermedad hepática terminal, genera morbilidad adicional y disminuye la calidad de vida de quienes la padecen.
En algunos casos es difícil hacer el diagnóstico de EH y definir su grado debido a la gran variabilidad de sus manifestaciones neurológicas, y a la similitud de estas con las de otras alteraciones que también se presentan con sintomatología neuropsiquiátrica.5
Por esta razón se requieren nuevos sistemas de clasificación y diagnóstico que permitan reconocer la EH desde las formas más leves y tempranas.6,7
A pesar de no existir un tratamiento generalmente aceptado para la EH, sus manifestaciones clínicas son reversibles con medidas como la dieta proteica y la terapia farmacológica.
PATOGÉNESIS
La patogénesis de la EH es multifactorial y al respecto se han planteado diferentes hipótesis que incluyen, entre otras, las siguientes: la neurotoxicidad del amonio, ácido gamma–amino–butírico (GABA)/benzodiacepinas, edema de los astrocitos, toxicidad por manganeso, opiáceos endógenos, trastornos de la neurotransmisión de histamina y serotonina y estrés oxidativo.1,2,5,8–11 De ellas, la más aceptada ha sido la neurotoxicidad del amonio que, al parecer, podría explicar gran parte de las demás.
El amonio
El amonio es una neurotoxina que, unida a factores aún no establecidos, podría precipitar el desarrollo de la EH.1,2,9 Las sustancias tóxicas intestinales, entre las que se incluye el amonio, se incrementan en la circulación debido a las derivaciones portosistémicas y a los defectos de la función hepática. Así mismo, la alta ingesta de proteínas de origen animal y los sangrados gastrointestinales pueden precipitar el aumento de esta sustancia en pacientes con alteración previa de la función hepática.9
En el cerebro, el amonio estimula la producción de glutamina y el consumo de ATP y glutamato (forma ionizada del ácido glutámico). En modelos experimentales se ha comprobado12–17 que los efectos anteriormente mencionados llevan a alteraciones en la permeabilidad de la barrera hematoencefálica, influyen sobre la neurotransmisión glutamatérgica e incrementan la expresión neuronal de la óxidonítrico sintetasa. Igualmente, se ha relacionado el amonio con disfunción en la neurotransmisión GABAérgica, incremento en la densidad de receptores benzodiacepínicos de tipo periférico (PTBR, por su sigla en inglés) y con anormalidades en los astrocitos.5,8,9,18
Los astrocitos
Los astrocitos se encargan de la detoxificación cerebral del amonio, porque son las únicas células cerebrales que contienen la enzima glutaminosintetasa, la cual cataliza el paso de amonio a glutamina.10 En la falla hepática aguda, el aumento rápido de la concentración intracelular de amonio en los astrocitos, en respuesta a su nivel elevado genera su edematización, lo cual puede ocasionar incremento del gradiente osmótico de la barrera hematoencefálica; como consecuencia pueden desarrollarse edema cerebral, aumento de la presión intracraneal e incluso la muerte por herniación cerebelosa.8,19,20
En pacientes con enfermedad hepática crónica, la EH se caracteriza neuropatológicamente por alteraciones morfológicas y funcionales de los astrocitos. Aunque puede ocurrir edema de tales células, por lo general es insuficiente para causar aumento en la presión intracraneal. Los cambios morfológicos característicos se conocen como astrocitosis tipo II del Alzheimer, en la que los astrocitos presentan núcleos largos e hinchados, nucléolos prominentes y marginación de la cromatina. Funcionalmente se encuentran alteraciones en la expresión de la proteína ácida fibrilar glial, en los transportadores de glutamato y en los receptores benzodiacepínicos de tipo periférico.11
GABA/benzodiacepinas
Investigaciones recientes sustentan la idea de que la activación en el sistema nervioso central de receptores benzodiacepínicos de tipo periférico contribuye a la patogénesis de las manifestaciones clínicas de la EH.21 Esto se ha demostrado por los hallazgos en tejidos cerebrales obtenidos en autopsias de pacientes con EH, en los que hay un incremento de los sitios de unión de los PTBR.22 La activación de los PTBR genera un incremento de la captación de colesterol, lo que a su vez aumenta la síntesis de neuroesteroides cerebrales que tienen propiedades moduladoras alostéricas positivas en el sistema inhibitorio GABA.22
Diferentes estudios han descrito la presencia de sustancias similares a las benzodiacepinas en el líquido cefalorraquídeo de pacientes con EH y en modelos animales de esta enfermedad.23–25 Dichas sustancias podrían contribuir al desarrollo de los problemas neurológicos asociados a la EH modulando los receptores GABA a través del sitio de unión de las benzodiacepinas. Entre estas sustancias, se han caracterizado algunos metabolitos de la hemoglobina como la hemina y la protoporfirina IX que pueden aumentar en la EH y acumularse en el sistema nervioso central donde producirían efectos GABAérgicos.26
CLASIFICACIÓN
La terminología que se utiliza actualmente para clasificar la EH es la propuesta en el Congreso Mundial de Gastroenterología en 1998.6 En la tabla n.º 1 se presenta dicha clasificación, que está basada en la naturaleza de la anormalidad hepática, así: es de tipo A cuando ocurre en pacientes con falla hepática aguda; de tipo B en pacientes con derivación portosistémica sin enfermedad hepatocelular intrínseca; y de tipo C en pacientes con cirrosis que además presentan hipertensión portal o derivación portosistémica.

En la EH de la enfermedad hepática crónica, se definieron las subcategorías episódica y persistente dependiendo de la duración y características de las manifestaciones neurológicas.
En la subcategoría persistente no desaparecen los déficits neuropsiquiátricos y, a pesar de las fluctuaciones en el nivel de conciencia, los pacientes no regresan al estado mental normal. Esta subcategoría también se subdivide en leve (EH grado 1), grave (EH grados 2–4) y dependiente del tratamiento cuando los síntomas se desarrollan luego de suspender la medicación. En el Congreso antes mencionado se propuso la subcategoría EH mínima para reemplazar el término EH subclínica.
DIAGNÓSTICO
El diagnóstico de EH es de exclusión porque también puede haber sintomatología neuropsiquiátrica en otros trastornos metabólicos, enfermedades infecciosas, accidentes vasculares intracraneales y lesiones cerebrales que ocupan espacio.5
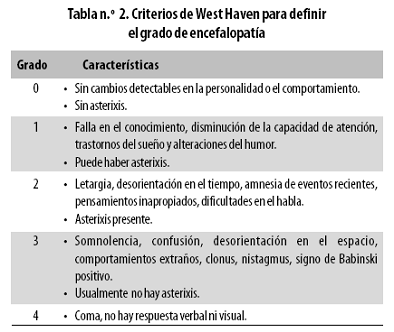
En el caso de la EH manifiesta, el diagnóstico requiere reconocer clínicamente el grado de encefalopatía, que actualmente se basa en los criterios de West Haven (Tabla n.º 2), tener conocimiento o sospecha de que el paciente presenta cirrosis, excluir otras enfermedades que cursan con manifestaciones similares o identificar factores precipitantes, entre ellos: hemorragias digestivas, infecciones, uso de sedantes, desequilibrio hidroelectrolítico, hematomas intracraneales, disfunción tiroidea, hipoglucemia, hipoxia, hipercapnia e intoxicaciones.1
El diagnóstico de la EH mínima27 es más complicado porque la evaluación neurológica es normal y los defectos cognitivos solo se pueden detectar mediante pruebas psicométricas como la de corrección de números, partes A y B, la del diseño de bloques y la de los símbolos digitales.6,28 Sin embargo, la mayoría de estas pruebas tienen derechos de autor que impiden su fácil acceso y hacerlas requiere mucho tiempo y la colaboración del paciente, además de disponer de personal entrenado para su interpretación. Por todo ello su disponibilidad es limitada. Otros estudios que pueden ser útiles para el diagnóstico de EH incluyen los potenciales evocados auditivos P300 y el electroencefalograma; no obstante, su alto costo los hace poco asequibles en la práctica clínica.29
La resonancia magnética (RM) cerebral también podría ser útil en el diagnóstico de la EH. En diferentes estudios30,31 se ha hallado que es posible observar mediante este examen una zona hiperintensa en los ganglios basales de pacientes con cirrosis, probablemente por depósito de manganeso. Este hallazgo, además, se correlaciona con el grado de derivación portosistémica que tenga el paciente, por lo cual se plantea que sería de gran utilidad para el diagnóstico de la EH de tipo B.
En los casos de EH por falla hepática aguda, la medición del nivel arterial de amonio puede ser útil para el pronóstico, porque cuando está por encima de 150 mg/dL se asocian con un riesgo alto de complicaciones y de herniación cerebelosa. A pesar de esto, los resultados de las mediciones del nivel de amonio son de poca utilidad para el diagnóstico de la EH en la enfermedad hepática crónica, pues no siempre se correlacionan.2,32
TRATAMIENTO
Se dispone de varias medidas que ayudan a controlar las manifestaciones clínicas de la EH, a mejorar la calidad de vida de los pacientes y a alargar la expectativa de la misma. Pese a esto no se puede olvidar que la principal medida terapéutica es identificar y tratar los factores que precipitaron el desarrollo de una EH manifiesta o mínima33,34 (Tabla n.º 3).

Dieta proteica
Durante muchos años la restricción proteica en la dieta se ha considerado como parte del tratamiento de la EH; se comienza con una ingesta proteica mínima (0,8–1 g/kg de peso);35 sin embargo, se ha demostrado que con este esquema de consumo proteico inicial excesivamente limitado se causa un balance nitrogenado negativo y se genera un hipercatabolismo proteico con aumento en la producción de amonio a expensas de la pérdida de músculo. La malnutrición proteica ocurre en 20–60% de los pacientes con cirrosis hepática y está asociada a complicaciones, entre ellas ascitis refractaria, hemorragia variceal y peritonitis bacteriana espontánea.36–39
En los informes de algunos estudios recientes se ha planteado que se puede permitir la ingesta de una cantidad normal de proteínas, de forma segura, a pacientes con episodios de EH;40,42 incluso se ha demostrado que con una dieta rica en proteínas y calorías se obtiene una resolución notoria de las manifestaciones clínicas características de este síndrome. Sin embargo, es necesario modificar el tipo de proteínas que van a consumir estos pacientes, es decir, deben ser principalmente de origen vegetal como las contenidas en las leguminosas; también las proteínas derivadas de los productos lácteos tienen efectos benéficos en los pacientes con EH; también es importante incluir en la dieta tubérculos y vegetales de hojas así como frutas, por su aporte de fibra que favorece el buen funcionamiento intestinal.
Disacáridos no absorbibles
La lactulosa y el lactitol son disacáridos no absorbibles con acción catártica, encargados de remover los sustratos amoniogénicos endógenos y los provenientes de la dieta. Estas sustancias son actualmente los principales agentes terapéuticos para los pacientes con EH crónica.34,40,43
La lactulosa disminuye el pH en el colon ocasionando una merma en la producción y absorción de amonio; de ello resulta un desplazamiento del amonio plasmático a la luz intestinal y por consiguiente un aumento de su excreción.44 La dosis diaria oscila entre 30 y 60 g y está sujeta a cambios según la cantidad, consistencia y pH de las heces; en un porcentaje alto de los pacientes hay efectos adversos como náuseas, flatulencia, diarrea y dolor abdominal.43 A pesar de su amplia difusión en el tratamiento de la EH, diversos ensayos clínicos no han encontrado un efecto significativo con el uso de lactulosa/lactitol comparado con el de placebo tanto en la reversibilidad de la encefalopatía como en su tasa de mortalidad,45 lo que ha llevado a concluir que estos compuestos fueron introducidos para el tratamiento de la EH sin tener una evidencia clínica apropiada; se está por lo tanto a la espera de nuevas evidencias que permitan dilucidar su verdadero papel en la terapia de esta enfermedad.
L–ornitina aspartato (OA)
Otra manera de reducir el nivel de amonio y mejorar el estado mental de los pacientes con EH es la administración de OA, que suministra los sustratos necesarios para el metabolismo del amonio llevando a la reducción de este en la sangre.34,43 Estudios clínicos han sugerido que la administración enteral o parenteral de OA tiene buen efecto terapéutico en pacientes cirróticos y con EH moderada;46,47 además, se ha encontrado que el tratamiento con OA estimula la detoxificación extrahepática del amonio,48 y tiene efectos directo e indirecto sobre la función hepática deteriorada de estos pacientes.
Antibióticos orales
La administración de antibióticos orales se usa ampliamente en pacientes con EH. Entre los más utilizados se encuentran la rifaximina y la neomicina; en algunos trabajos se ha definido que la eficacia de esta última es similar a la de la lactulosa.43,49 Por otra parte, se ha demostrado que la rifaximina a la dosis de 1.200 mg/día tiene mayor efecto sobre la disminución del nivel plasmático de amonio en pacientes con EH que el producido por los disacáridos no absorbibles. En investigaciones recientes se ha demostrado que con el uso de rifaximina en pacientes con EH aguda o crónica se logra un mayor porcentaje de mejoramiento frente a los que son tratados con lactulosa o lactitol, e incluso, que la mejoría se presenta en un intervalo más corto;50,51 además, la tolerancia es mejor y el índice de efectos adversos mucho menor en pacientes tratados con rifaximina.49,52 Sin embargo, no se la recomienda como terapia preventiva de la EH después de una derivación intrahepática portosistémica.52
Agentes neuromoduladores
Por otra parte se cuenta con los antagonistas de los receptores benzodiacepínicos como el flumazenil. Al evaluar su eficacia en el tratamiento de la EH, se estableció en diferentes estudios que, en comparación con grupos placebo, se obtiene un resultado favorable independientemente de la presencia de benzodiacepinas exógenas;53 no obstante, se encontró que el beneficio es temporal y que es más marcado en la encefalopatía aguda. De igual manera, la mejoría clínica del grado de encefalopatía no es completa, por lo que no se la recomienda como tratamiento clínico de rutina.34
REFERENCIAS BIBLIOGRÁFICAS
1. Mullen KD. Pathogenesis, clinical manifestations, and diagnosis of hepatic encephalopathy. Semin Liver Dis 2007; 27 (Suppl. 2): 3–9. [ Links ]
2. Mullen KD, Ferenci P, Bass NM, Leevy CB, Keeffe EB. An algorithm for the management of hepatic encephalopathy. Semin Liver Dis 2007; 27 (Suppl. 2): 32–47. [ Links ]
3. Stewart CA, Malinchoc M, Kim WR, Kamath P. Hepatic encephalopathy as a predictor of survival in patients with end–stage liver disease. Liver Transpl 2007; 13: 1366–1371. [ Links ]
4. Kamath PS, Wiesner RH, Malinchoe M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end–stage liver disease. Hepatology 2001: 33: 464–470. [ Links ]
5. Blei AT, Cordoba J. The Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol 2001; 96: 1968–1976. [ Links ]
6. Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy –definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congress of Gastroenterology, Vienna, 1998. Hepatology 2002; 35: 716–721. [ Links ]
7. Kircheis G, Fleig WE, Gortelmeyer R, Grafe S, Haussinger D. Assessment of low–grade hepatic encephalopathy: A critical analysis. J Hepatol 2007; 47: 642–650. [ Links ]
8. Ong JP, Mullen K. Hepatic encephalopathy. Eur J Gastroenterol Hepatol 2001; 13: 325–334. [ Links ]
9. Katayama K. Ammonia metabolism and hepatic encephalopathy. Hepatol Res 2004; 30S: S71–S78. [ Links ]
10. Haussinger D, Schliess F, Kircheis G. Conference Proceedings. Pathogenesis of hepatic encephalopathy. J Gastroenterol Hepatol 2002; 17: S256–S259. [ Links ]
11. Butterworth R. Pathophysiology of hepatic encephalopathy: A new look at ammonia. Metab Brain Dis 2002; 17: 221–227. [ Links ]
12. Blei AT. Hepatic encephalopathy. In: Bircher J, Benhamou JP, McIntyre N, Rizzeto M, Rodés J, eds. Oxford Textbook of Hepatology. Oxford, UK: Oxford University Press; 1999:765–786. [ Links ]
13. Butterworth RE, Giguere JF, Michaud J, Lavoie J, Layrargues GP. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol 1987; 6: 1–12. [ Links ]
14. Vogels BA, Maas MA, Daalhuisen J, Quack G, Chamuleau RA. Memantine, a noncompetitive NMDA receptor antagonist proves hyperammonemia–induced encephalopathy and acute hepatic encephalopathy in rats. Hepatology 1997; 25: 820–827. [ Links ]
15. Butterworth R. Hepatic encephalopathy and brain edema in acute hepatic failure: does glutamate play a role? [Editorial comment]. Hepatology 1997; 25: 1032–1034. [ Links ]
16. Norenberg MD. Astroglial dysfunction in hepatic encephalopathy. Metab Brain Dis 1998; 13: 319–335. [ Links ]
17. Rao VL, Butterworth RE. Neuronal nitric oxide synthase and hepatic encephalopathy. Metab Brain Dis 1998; 13: 175–189. [ Links ]
18. Aguila–Mansilla N, Bender AS, Norenberg MD. Effect of ammonia, peripheral benzodiazepine ligands and neurosteroids on GABA uptake in cultured astrocytes. J Neurochem 1998; 70 (Suppl.) IS28. [ Links ]
19. Blei AT, Larsen FS. Pathophysiology of cerebral edema in fulminant hepatic failure. J Hepatol 1999; 31: 771–776. [ Links ]
20. Ahboucha S, Pomier–Layrargues G, Butterworth R. Increased brain concentrations of endogenous (nonbenzodiazepine) GABA–A receptor ligands in human hepatic encephalopathy. Metab Brain Dis 2004; 19: 241–251. [ Links ]
21. Cagnin A, Taylor–Robinson SD, Forton DM, Banati RB. In vivo imaging of cerebral 'peripheral benzodiazepine binding sites' in patients with hepatic encephalopathy. Gut 2006; 55: 547–553. [ Links ]
22. Butterworth RF. The astrocytic ('peripheral–type') benzodiazepine receptor: role in the pathogenesis of portal–systemic encephalopathy. Neurochem Int 2000; 36: 411–416. [ Links ]
23. Olasmaa M, Rothestein JD, Guidotti A, Weber RJ, Paul SM, Spector S, et al. Endogenous benzodiazepine receptor ligands in human and animal hepatic encephalopathy. J Neurochem 1990; 55: 2015–2023. [ Links ]
24. Basile AS, Panell L, Jaouni T, Gammal SH, Fales HM, Jones EA, et al. Brain concentrations of benzodiazepines are elevated in an animal model of hepatic encephalopathy. Proc Natl Acad Sci USA 1990; 87: 5263–5267. [ Links ]
25. Gammal SH, Basile AS, Geller D, Skolnick P, Jones EA. Reversal of the behavioral and electrophysiological abnormalities of an animal model of hepatic encephalopathy by benzodiazepine receptor ligands. Hepatology 1990; 11: 371–378. [ Links ]
26. Ruscito B, Harrison N. Hemoglobin metabolites mimic benzodiazepines and are possible mediators of hepatic encephalopathy. Blood 2003; 102: 1525–1528. [ Links ]
27. Amodio P, Montagnese S, Gatta A, Morgan MY. Characteristics of minimal hepatic encephalopathy. Metab Brain Dis 2004; 19: 253–267. [ Links ]
28. Amodio P, Del Piccolo F, Marchetti P, Angeli P, Iemmolo R, Caregaro L, et al. Clinical features and survival of cirrhotic patients with subclinical cognitive alterations detected by the number connection test and computerized psychometric tests. Hepatology 1999; 29: 1662–1667. [ Links ]
29. Lockwood AH. Clinical and laboratory features of hepatic encephalopathy. In: Hepatic Encephalopathy. Boston: Butterworth–Heinemann; 1992: 2–31. [ Links ]
30. Butterworth R. Pathogenesis of hepatic encephalopathy: new insights from neuroimaging and molecular studies. J Hepatol 2003; 39: 278.285. [ Links ]
31. Mullen KID, Cole M, Foley JM. Neurological deficits in 'awake' cirrhotic patients on hepatic encephalopathy treatment: missed metabolic or metal disorder? [Editorial comment]. Gastroenterology 1996; 111: 256–257. [ Links ]
32. Lockwood AH. Blood ammonia levels and hepatic encephalopathy. Metab Brain Dis 2004; 19: 345–349. [ Links ]
33. Jalan R, Hayes PC. Hepatic encephalopathy and ascites. Lancet 1997; 350: 1309–1315. [ Links ]
34. Kircheis G, Häussinger D. Management of hepatic encephalopathy. J Gastroenterol Hepatol 2002; 17; S260–S267. [ Links ]
35. Uribe M, Conn HO. Dietary management of portalsystemic encephalopathy. In: Conn HO, Bircher J, eds. Hepatic encephalopathy: syndromes and therapies. Bloomington, Ill.: Medi–Ed Press; 1994: 331–349. [ Links ]
36. Kalman DR, Saltzman JR. Nutrition status predicts survival in cirrhosis. Nutr Rev 1996; 54: 217–219. [ Links ]
37. Nutritional status in cirrhosis: Italian Multicentre Cooperative Project on Nutrition in Liver Cirrhosis. J Hepatol 1994; 21: 317–325. [ Links ]
38. Lautz HU, Selberg O, Korber J, Burger M, Muller MJ. Protein–calorie malnutrition in liver cirrhosis. Clin Investig 1992; 70: 478–486. [ Links ]
39. Merli M, Riggio O, Dally L, and PINC (Policentrica Italiana Nutrizione Cirrosi). Does malnutrition affect survival in cirrhosis? Hepatology 1996; 23: 1041–1046. [ Links ]
40. Shawcross D, Jalan R. Dispelling myths in the treatment of hepatic encephalopathy. Institute of Hepatology, University College London, London. Lancet 2005; 365: 431–433. [ Links ]
41. Córdoba J, López–Hellín J, Planas M, Sabín P, Sanpedro F, Castro F, et al. Normal protein diet for episodic hepatic encephalopathy: results of a randomized study. J Hepatol 2004; 41: 38–43. [ Links ]
42. Gheorghe L, Iacob R, Vãdan R, Iacob S, Gheorghe I. Improvement of hepatic encephalopathy using a modified high–calorie high–protein diet. Rom J Gastroenterol 2005; 14: 231–238. [ Links ]
43. Riordan SM, Williams R. Treatment of hepatic encephalopathy. New Engl J Med 1997; 337: 473–479. [ Links ]
44. Conn HO, Lieberthal MM. The hepatic coma syndromes and lactulose. Baltimore: Williams & Wilkins, 1979. [ Links ]
45. Als–Nielsen B, Gluud LL, Gluud C. Non–absorbable disaccharides for hepatic encephalopathy: systematic review of randomised trials. BMJ 2004; 328; 1046–1053. [ Links ]
46. Kircheis G, Quack G, Erbler H. L–ornithine–L–aspartate in the treatment of hyperammonemia and hepatic encephalopathy. In: Conn HO, Bircher J, eds. Hepatic encephalopathy: syndromes and therapies. Bloomington, Ill.: Medi–Ed Press, 1994: 373–383. [ Links ]
47. Herlong HF, Maddrey WC, Walser M. The use of ornithine salts of branched–chain ketoacids in portal–systemic encephalopathy. Ann Intern Med 1980; 93: 545–550. [ Links ]
48. Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L–ornithine–L–aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30: 636–640. [ Links ]
49. Bass NM. Emerging therapies for the management of hepatic encephalopathy. Semin Liver Dis 2007; 27 (Suppl. 2): 18–25. [ Links ]
51. Williams R, James OFW, Warnes TW, Morgan MY. Evaluation of the efficacy and safety of rifaximin in the treatment of hepatic encephalopathy: a double–blind, randomized, dose–finding multicentre study. Eur J Gastroenterol Hepatol 2000; 12: 203–208. [ Links ]
52. Baker DE. Rifaximin: A nonabsorbed oral antibiotic. Rev Gastroenterol Disord 2005; 5: 19–30. [ Links ]
53. Goulenok C, Bernard B, Cadranel JF, Thabut D, Di Martino V, Opolon P, et al. Flumazenil vs. placebo in hepatic encephalopathy in patients with cirrhosis: a metaanalysis. Aliment Pharmacol Ther 2002; 16: 361–372. [ Links ]
Recibido: junio 23 de 2008
Aceptado: agosto 30 de 2008

