










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkIatreia
Print version ISSN 0121-0793
Iatreia vol.22 no.1 Medellín Jan./Mar. 2009
ARTÍCULO DE REVISIÓN
Síndromes de Bartter y Gitelman: revisión de los aspectos genéticos, fisiopatológicos y clínicos
Bartter and Gitelman syndromes: a review of genetic, physiopathological and clinical aspects
Lina María Serna Higuita1; Lina María Betancur Londoño1; Carlos Mauricio Medina Vásquez1; Nicolás Pineda Trujillo2; Juan José Vanegas Ruiz3
1 Residente de Pediatría, Universidad Pontificia Bolivariana, Medellín, Colombia.
2 MSc, PhD en Genética, Grupo de Mapeo Genético, Departamento de Pediatría y Puericultura, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
3 Nefrólogo infantil. Facultad de Medicina, Universidad Pontificia Bolivariana. Departamento de Pediatría y Puericultura, Facultad de Medicina, Universidad de Antioquia.
Dirección de contacto: linaserna77@hotmail.com
Resumen
Los síndromes de Bartter y Gitelman son trastornos hereditarios caracterizados por una reducción marcada del transporte de sales por el asa ascendente gruesa de Henle. Los pacientes con el síndrome de Bartter presentan grandes pérdidas renales de agua, hipotensión arterial, alcalosis metabólica, hipokalemia e hipercalciuria y tienen un riesgo alto de sufrir nefrolitiasis. Estudios recientes han permitido subdividir el síndrome de Bartter en cinco tipos de acuerdo con el defecto genético y el fenotipo. El tipo 1 es causado por una mutación en el gen que codifica para el cotransportador cloro–sodio–potasio; el tipo 2 se debe a una mutación en el gen que codifica para el canal de potasio ROMK. Estas dos variantes genéticas se denominan conjuntamente síndrome de Bartter neonatal por el comienzo temprano de los síntomas, con polihidramnios materno, prematuridad, poliuria grave y nivel elevado de prostaglandina E2. El tipo 3, conocido como síndrome de Bartter clásico, es causado por mutaciones en el gen que codifica para el canal de cloro CLC–Kb; se detecta desde la niñez con retardo del crecimiento, poliuria, polidipsia y anorexia. El tipo 4 se asocia a sordera neurosensorial y se caracteriza por una mutación en el gen que codifica para la proteína Barttin la cual hace parte de los canales de cloro CLC–Kb y CLC–Ka; el tipo 5 se debe a una mutación en el gen que codifica para un receptor de calcio ubicado en la membrana basolateral del asa ascendente de Henle; estos pacientes, además de los síntomas comunes a los de otros tipos, presentan déficit de paratohormona. El síndrome de Gitelman tiene un fenotipo más leve y una presentación más tardía que el de Bartter; se diferencia de este porque los pacientes tienen hipomagnesemia e hipocalciuria; pueden ser asintomáticos o presentar debilidad muscular transitoria, parestesias, parálisis e incluso alteraciones del ritmo cardíaco. Los avances en genética molecular han permitido la clasificación adecuada de estos síndromes y han abierto puertas para diferentes opciones terapéuticas. Esta revisión incluye aspectos genéticos, fisiopatológicos y clínicos de estos síndromes.
Palabras clave: Alcalosis metabólica hipokalémica, Síndrome de Bartter, Síndrome de Gitelman
Summary
Bartter and Gitelman syndromes are hereditary disorders characterized by a remarkable reduction of salt transportation by the thick ascending limb of the Henle's loop. Consequently, patients suffering from Bartter syndrome present with renal salt wasting, low blood pressure, hypokalemic metabolic alkalosis and hypercalciuria, and are at risk of developing renal stones. Based on recent studies, the Bartter syndrome has been subdivided into five types according to the genetic defect involved and the phenotype: type 1 is caused by a mutation in the gene coding for the chloride, sodium, and potassium co–transporter; type 2 is due to a mutation in the gene coding for the ROMK potassium channel. These two genetic variations are jointly denominated neonatal Bartter syndrome, because of their early clinical presentation, with maternal polyhydramnios, prematurity, severe polyuria and high levels of E2 prostaglandine. Type 3, also known as classic Bartter syndrome, is produced by mutations in the gen that codes for the chloride CLC–Kb channel; this type is detected since childhood with growth delay, polyuria, polydipsia and anorexia. Type 4, which is associated with neurosensorial deafness, is characterized by a mutation in the gen that codes for the Barttin protein which is a part of the CLC–Kb and CLC–Ka chloride channels. Type 5 appears because of a mutation in the gene that codes for a calcium receptor located at the basolateral membrane of the ascending limb of Henle's loop; patients with this type develop parathormone deficit, as well as the symptoms that are common to all types of the syndrome. The phenotype of Gitelman syndrome is less severe and its clinical presentation is delayed; it differs from the Bartter syndrome in that patients have hypomagnesemia and hipocalciuria. They may be asymptomatic or show transitory muscular weakness, paresthesias, paralysis and even cardiac rhythm alterations. Recent advances in molecular genetics have made it possible to distinguish between the different clinical types and are the basis for several therapeutic options. This review includes genetic, physiopathological and clinical aspects of the Bartter and Gitelman syndromes.
Key words: Bartter syndrome, Gitelman syndrome, Hipokalemic metabolic alkalosis
INTRODUCCIÓN
Los síndromes de Bartter y Gitelman son tubulopatías hereditarias, estrechamente relacionadas con transmisión de tipo mendeliano, en las que hay deterioro de los mecanismos de concentración de la orina y transporte del cloruro de sodio (NaCl) en la nefrona distal.1,2 Los pacientes con estos síndromes comparten algunas características clínicas que incluyen: pérdida renal de sales, alcalosis metabólica hipokalémica,3 hiperaldosteronismo hiperreninémico, presión arterial normal e hiperplasia del aparato yuxtaglomerular.1,2,4 El síndrome de Bartter se clasifica en varios tipos que difieren en cuanto a la edad de comienzo, la gravedad de los síntomas, la pérdida de electrólitos y la excreción urinaria de algunas prostaglandinas.5 El síndrome de Gitelman se diferencia del de Bartter porque en él se presentan hipomagnesemia e hipocalciuria.4,6
En este artículo se presenta una revisión de los hallazgos más recientes de estos dos síndromes con énfasis en aspectos genéticos y fisiopatológicos. Además se describen la caracterización clínica de cada síndrome, el diagnóstico y el tratamiento.
EPIDEMIOLOGÍA
Estos síndromes son infrecuentes y en la literatura médica se encuentran pocos informes al respecto; hasta la fecha no hay datos sobre su frecuencia en Colombia; un estudio publicado en 1988 informó una prevalencia aproximada de 1,2/1.000.000 de habitantes en Goteborg, Suecia.7 Los informes iniciales fueron en personas de raza negra pero no existe una predilección étnica y, en cuanto al sexo, afecta por igual a hombres y mujeres.8
ETIOLOGÍA
El transporte renal del ion cloro es un proceso complejo que involucra una adecuada coordinación entre diferentes canales luminales: canal NCCK2, canal rectificador de potasio (ROMK), canal basolateral de cloro (CLC–Kb/Barttin), así como otros cotransportadores y canales.9 El cloro se reabsorbe en la membrana luminal del asa ascendente gruesa de Henle, por medio de un cotransporte con el potasio a través del canal NaK2Cl,10 que tiene gran capacidad de transporte pasivo de sodio, lo que contribuye a la reabsorción de cloro; este cotransporte pasivo es facilitado por la baja concentración celular de sodio y cloro que salen al compartimiento sanguíneo por medio de la bomba Na–K ATPasa y el canal CLC–Kb/Barttin, ambos ubicados en la membrana basolateral. Asociado a los mecanismos anteriores, el canal ROMK se expresa en la membrana luminal del asa ascendente de Henle, aumenta la funcionalidad del canal NaK2Cl, realizando un reciclaje apical permanente del potasio, o sea, permitiendo la entrada de este ion de la luz del túbulo a la célula.9–11 De esta manera se suple el potasio luminal necesario para su función de cotransporte;12 el potasio apical genera un gradiente positivo en la luz, aumentando la reabsorción paracelular de una fracción significativa de sodio, potasio y magnesio9,13 (Figura n.º 1).
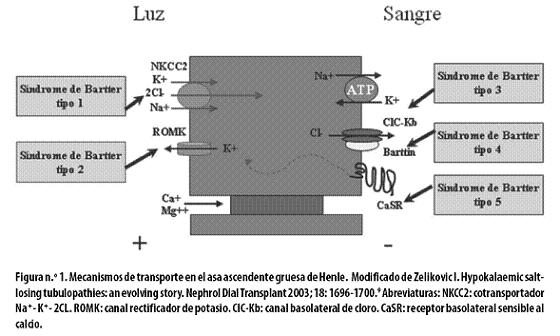
El canal ROMK ayuda a la secreción de potasio en la nefrona distal. El transporte de cloro del túbulo contorneado distal es mediado por el cotransportador de NaCl sensible a tiazidas ubicado en la membrana luminal9 (Figura n.º 2).
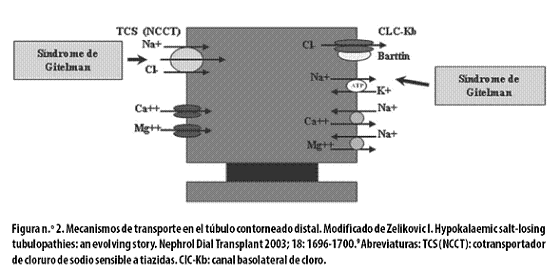
Las diferentes mutaciones en cualquiera de estos canales producen cuadros clínicos muy bien definidos, entre los cuales se encuentran los síndromes de Bartter y Gitelman.13
GENÉTICA Y PRESENTACIÓN CLÍNICA
Síndrome de Bartter
Clasificación
Se han descrito 5 tipos del síndrome de Bartter, dependiendo del canal alterado:
1. Bartter tipo 1: también se lo denomina, junto con el tipo 2, síndrome de Bartter neonatal y era conocido anteriormente como síndrome de hiperprostaglandina E. Se debe a una mutación en el gen NKCC2 (SLC12A1)14 el cual reside en el cromosoma 15q15–21;9,12 esta variante genética se caracteriza por sintomatología prenatal; su fenotipo y anormalidades bioquímicas son similares a los inducidos por la terapia crónica con furosemida.9 El deterioro en el transporte de NaCl en el asa ascendente de Henle da lugar a aumento de la expresión de COX2, el que a su vez incrementa la síntesis de prostaglandina E, seguida por secreción de renina;15 por eso los pacientes con este síndrome se benefician del tratamiento con inhibidores de la COX2; al normalizar la excreción de prostaglandina E2 y la actividad de la renina plasmática disminuye hasta en un 50% la pérdida de sodio y agua.15
En un estudio realizado por Starremans y colaboradores13 se encontraron diferentes mutaciones en el gen NKCC2, a saber:
• Mutación missense en G193R, A267S y G319R.
• Mutación 526N que es una deleción del aminoácido asparagina en la posición 526.
• Mutación Y998X que genera un codón de parada prematura y una proteína truncada.
En la evaluación funcional del NKCC2 se encuentra que las mutaciones codifican para una proteína estable y bien ubicada pero no funcional, lo que genera una merma en la reabsorción de sales en el asa ascendente gruesa de Henle y un deterioro en el transporte transepitelial de calcio.9 Además, se han reportado otras mutaciones nonsense (N117X) y mutaciones de cambio en el marco de lectura (E792 y N984).16 La presentación clínica de este tipo se describirá más adelante.
2. Bartter tipo 2: también denominado, junto con el tipo 1, síndrome de Bartter neonatal; se caracteriza por un defecto en el canal ROMK, causado por una mutación en el gen KCNJ1,17 ubicado en el cromosoma 11q24–25.18 Este gen codifica para un transportador de membrana para el paso del potasio;19 su mutación genera un deterioro en la secreción de potasio en el asa de Henle y en el túbulo colector.9,11
Todas las mutaciones reportadas hasta la fecha se localizan en el exón 5 del gen ROMK y comprenden sustitución de pares de bases, codones de parada prematura e inserciones, que causan errores en la lectura del gen.12 La distribución de estas mutaciones dentro de la proteína (regiones N terminal, central y C terminal) se asocia con los diferentes mecanismos fisiopatológicos encontrados, así:
1. Alteración en los componentes del poro que deteriora el paso de potasio.
2. Defectos en la regulación del canal ROMK.
3. Alteraciones en el transporte a la superficie celular.10
Además, algunas mutaciones producen una proteína truncada. En un estudio se encontraron cuatro grupos diferentes de mutaciones del canal ROMK, por medio de cultivo en oocitos de Xenopus, análisis por inmunofluorescencia, estudio de voltaje y análisis de Western blot.10 El primer grupo comprende las mutaciones F95S, A177T, R311W, Arg338Stop,12 que generan una proteína no funcional, debido a cambios en la formación del poro del canal. El segundo consiste en las mutaciones V72E, D74Y, D108H, V122E, G167E, Y314C, L320P Y F325C; este grupo muestra un defecto en la funcionalidad de la proteína y alteración de su transporte a la membrana plasmática. El tercero comprende las mutaciones T71M, A306T y R324L en las que se afecta el transporte de la proteína pero no su funcionalidad. El cuarto grupo se compone de dos mutaciones que generan los codones de parada W77X, Y79X.10
El síndrome de Bartter neonatal (tipos 1 y 2) amenaza la vida porque produce alcalosis hipokalémica grave. Clínicamente se caracteriza por polihidramnios y parto prematuro,5,20 generalmente entre las semanas 24 y 30 de la gestación;13 el incremento del líquido amniótico es consecuencia de la poliuria fetal, que persiste en el período posnatal y que, asociada a la hipostenuria (baja densidad urinaria) de estos pacientes, les ocasiona pérdidas graves de agua y sal; también presentan alcalosis metabólica hipokalémica, expresión elevada de prostaglandina E2 y nivel alto de renina (causado por la disminución del volumen vascular que activa el eje renina–aldosterona) e hipercalciuria grave con nefrocalcinosis temprana que los puede llevar a insuficiencia renal.4,6,9 Pueden presentar además osteopenia y marcado retardo del crecimiento.12,13,19 Algunas manifestaciones sistémicas son fiebre, vómito y, ocasionalmente, diarrea, debidos a un marcado estímulo de la producción renal y sistémica de prostaglandina E2.12,21 Se han reportado pacientes con retardo mental, pero este es prevenible si se hacen tempranamente el diagnóstico y el tratamiento.
En los pacientes con Bartter tipo 2 se pueden encontrar ocasionalmente una hiperkalemia transitoria y acidosis antes de la alcalosis hipokalémica típica.21
3. Bartter tipo 3: también se lo conoce como síndrome de Bartter clásico y se observa principalmente en la infancia;2,6 la mutación responsable se ubica en el gen que codifica para el canal de cloro CLC–Kb (CLCNKB), situado en el cromosoma 1p36;2,6,22 se han descrito al menos 26 mutaciones diferentes, con una alta variabilidad fenotípica, por lo que puede, incluso, simular el síndrome de Bartter neonatal o el síndrome de Gitelman con hipocalciuria e hipomagnesemia.23,24 En un estudio llevado a cabo en España en 10 pacientes no relacionados se encontró que eran homocigotos para una mutación missensse (A204T) en el gen CLC–Kb la cual posiblemente tiene un efecto fundador.2 También se ha encontrado una mutación nonsense, la W610X, que genera una proteína truncada con pérdida de la función. Esta mutación fue reportada en seis familias japonesas no relacionadas y nunca se la ha detectado en otras razas, lo que sugiere un ancestro común en esta población.25
La familia de los canales de cloro, CLC, está representada por nueve miembros en las membranas plasmáticas y en las organelas intracelulares,23,26 cada uno con 12 dominios transmembrana; sus funciones principales son: regulación del volumen celular, transporte de cloro a través de las células epiteliales tubulares del intersticio en el asa ascendente gruesa de Henle y en el túbulo contorneado distal y regulación de la excitabilidad eléctrica;2 los dos miembros, CLC–Ka y CLC–Kb, se expresan principalmente en el riñón en el asa ascendente de Henle (solo el CLC–Ka) y en las células intercaladas de los túbulos colector y distal,23 y son fundamentales para los mecanismos de concentración urinaria.26
Clínicamente, este tipo del síndrome se detecta desde la niñez, generalmente antes de los dos años;4 estos pacientes presentan una alcalosis metabólica hipokalémica con normocalciuria o hipercalciuria; sus síntomas consisten en poliuria, que puede manifestarse como enuresis, polidipsia, vómito, constipación, deseo de comer sal, retardo del crecimiento, fatiga y tendencia a la deshidratación; algunos pacientes pueden presentar parestesias, debilidad muscular y parálisis transitorias;27,28 la hipercalciuria en algunos casos puede llevarlos a nefrocalcinosis;29,30 sin embargo, la presión arterial es normal o baja.2,31 Estos pacientes, aunque tengan la misma mutación, pueden presentar expresiones fenotípicas diferentes incluso similares a las de otras enfermedades como el síndrome de Fanconi y la acidosis tubular renal; posiblemente ello se deba a diferencias interindividuales en la capacidad para compensar un defecto del transporte de cloro, o a la activación de rutas alternas para el flujo de cloro tales como el cotransportador K–Cl u otros canales de cloro.6,32,33
4. Bartter tipo 4: está asociado a sordera neurosensorial6 y se debe a una mutación en el gen que codifica para la proteína llamada Barttin (BSND),34,35 ubicado en el locus 1p31.4,23,36 La proteína Barttin es la subunidad b de los canales de cloro CLC–Ka y CLC–Kb y se requiere para su funcionalidad y transporte a la membrana plasmática;23,37,33 estos canales se ubican en la membrana basolateral de los túbulos renales y en el epitelio secretor de potasio del oído interno.4,9,33 La mutación en este gen produce incapacidad para la secreción de potasio dentro de la endolinfa causando sordera de tipo neurosensorial.23
Se han encontrado aproximadamente nueve mutaciones en este gen, así: tres debidas a la sustitución de aminoácidos (G10S, R8W, R8L),3,38 cuatro que generan pérdida en los codones de inicio, una en la que hay cambio de un aminoácido por otro (glicina por arginina en el codón 47 G47R),38 que codifica la región del exón 1, generando un deterioro del transporte transepitelial del canal de cloro CLC–Kb;34 y una mutación Q32X que produce una proteína truncada con solo 31 aminoácidos de un total de 320.34
Recientemente se informó de un paciente con síndrome de Bartter neonatal y sordera neurosensorial (Bartter tipo 3) que no presentaba ninguna mutación en el gen que codifica para la proteína Barttin, pero sí tenía mutaciones heterocigotas en los genes CLCNKA y CLCNKB; la pérdida de la función de los alelos de ambos canales genera un fenotipo grave del síndrome de Bartter con sordera neurosensorial muy similar a lo que pasa en la interacción de la proteína Barttin con ambos canales CLCNK.39 Los pacientes con Bartter tipo 4 se caracterizan por presentar un cuadro más grave que los afectados por los tipos 1 y 2 del síndrome: son prematuros debido al polihidramnios;37 al nacer tienen pérdida grave de sales por lo que requieren reemplazo con líquidos parenterales por períodos prolongados; también se observan en ellos hipokalemia, hipocalcemia, hipomagnesemia, hipercalciuria, retardo del desarrollo y marcada hipotonía. Sus facies son dismórficas con rasgos como cara triangular, frente prominente, ojos grandes y orejas que protruyen;4 algunos pueden presentar nefrocalcinosis y frecuentemente evolucionan a la falla renal crónica.36 La respuesta a la indometacina y al nimesulide es mala.6,23,36
5. Bartter tipo 5: la herencia de este tipo del síndrome es autosómica dominante; se debe a una mutación en el gen CASR que codifica para el receptor del calcio localizado en la membrana basolateral de las células del asa ascendente gruesa de Henle;4 la activación de este receptor produce un efecto inhibitorio en la reabsorción de NaCl así como en el transporte de los cationes divalentes a lo largo del asa.26 Este subtipo se caracteriza por hipocalcemia, déficit de paratohormona, hipokalemia, hipomagnesemia y nefrocalcinosis.34
SINDROME DE GITELMAN
Este síndrome, descrito por Gitelman en 1966 (citado por Konrad y Weber6 y por Reinalter y colaboradores40) es una forma diferente de alcalosis metabólica con hipomagnesemia e hipocalciuria;18,41,42 usualmente se inicia en la edad escolar pero puede diagnosticarse también en la vida adulta.4,5 Su prevalencia estimada es de 1/1.000.000 de habitantes, pero esto depende de la consanguinidad.43 Es una forma menos grave que el síndrome de Bartter porque se conserva la capacidad de concentrar la orina6,44 y no hay retardo del crecimiento ni poliuria; la sintomatología comienza a una edad relativamente tardía, generalmente por encima de los seis años; sin embargo, aunque se lo considera benigno, la combinación de hipokalemia con hipomagnesemia puede prolongar el intervalo QT y desencadenar arritmias que, como la taquicardia ventricular, pueden menazar la vida del paciente.43 Se ha informado que hasta el 50% de los pacientes con síndrome de Gitelman tienen prolongación del intervalo QT.45
Estos pacientes presentan debilidad muscular, fatiga, vértigo, polidipsia, nicturia, palpitaciones, presión arterial baja y crisis de tetania como el espasmo carpopedal;43 algunos de ellos desarrollan condrocalcinosis, posiblemente secundaria a la hipomagnesemia crónica. Otros síntomas menos frecuentes son: lipotimia, poliuria, artralgias, vómito, constipación, enuresis y parálisis periódicas.41,44 Su forma de herencia es autosómica recesiva aunque la mayoría de los pacientes son heterocigotos compuestos para diferentes mutaciones dentro del mismo gen, lo que puede explicar la gran variabilidad fenotípica que se observa en cuanto a la edad de comienzo y la gravedad de los síntomas.41
Este síndrome se debe a una mutación en el gen ubicado en el brazo largo del cromosoma 16, locus 16q13, codificado por 26 exones.9,41 Este gen (NCCT) codifica para el cotransportador de NaCl sensible a diuréticos tiazídicos, SLC12A3, 46 proteína de 1.021 aminoácidos que se expresa en la membrana apical de las células del túbulo contorneado distal.43,46,47
La hipocalciuria se explica por la disminución en la entrada de cloruro de sodio en el túbulo contorneado distal, lo que lleva a hiperpolarización celular, incrementando la reabsorción de calcio mediada a nivel apical por un canal de calcio epitelial y la salida basolateral por un intercambiador de Na/Ca2;9 aún no está clara la causa de la hipomagnesemia; se cree que es causada por un incremento del voltaje negativo en la luz lo que favorecería la secreción paracelular de magnesio y potasio.6 Otra hipótesis es que el bloqueo del NCCT genera un incremento en la apoptosis de las células del túbulo contorneado distal; sin embargo, se requiere más investigación en este campo.9
DIAGNÓSTICO
Síndrome de Bartter neonatal (tipos 1 y 2)
En estos dos subtipos el diagnóstico se puede hacer en el período prenatal por el alto contenido de cloro en el líquido amniótico,48,49 con niveles normales de sodio, potasio, calcio y prostaglandina E2. Sin embargo, la forma definitiva de establecer un diagnóstico es evaluar para la mutación en ADN extraído del cultivo de amniocitos obtenidos por amniocentesis.18
Los siguientes son hallazgos importantes en los exámenes de laboratorio de estos pacientes: densidad urinaria disminuida; niveles urinarios aumentados de calcio, sodio, cloro y potasio; alcalosis metabólica con hipokalemia. Otro hallazgo característico del síndrome de Bartter es la hiperreninemia, que en muchos casos está acompañada por aumento en los niveles de aldosterona y prostaglandina E.34
Síndrome de Bartter clásico (tipo 3)
En este caso el diagnóstico se hace al encontrar hipokalemia en el rango de 1,5 a 2,5 mEq/L, acompañada de hipocloremia, alcalosis metabólica y, en algunos casos, hipomagnesemia. Los hallazgos urinarios más característicos son: aumento en la fracción de excreción de potasio, sodio, cloro y calcio (aunque este último puede estar normal). Además estos pacientes no pierden completamente la capacidad de concentrar la orina lo que los diferencia de los que sufren los tipos 1 y 2.
Síndrome de Gitelman
En este síndrome se observa una marcada alcalosis metabólica (pH mayor de 7,45 y bicarbonato por encima de 29 mEq/L), con profunda hipokalemia (menos de 3 mEq/L), hipomagnesemia (por debajo de 0,5 mEq/L) e hipocalciuria (menos de 2 mg/kg/día). Estos pacientes presentan los mismos hallazgos de laboratorio que los del síndrome de Bartter pero se diferencian por presentar hipomagnesemia e hipocalciuria18 y por preservar la capacidad para concentrar la orina.
Un test de hidroclorotiazida con incremento máximo de la fracción excretada de cloro (AFECl) menor de 2,3% tiene buenas sensibilidad y especificidad para diagnosticar este síndrome.50 La biopsia renal, aunque demuestra la hiperplasia del aparato yuxtaglomerular, no es necesaria para confirmar estos síndromes.41
TRATAMIENTO
Síndrome de Bartter neonatal (tipos 1 y 2)
Al nacer el niño, el esfuerzo terapéutico se debe orientar a corregir la deshidratación y el desequilibrio hidroelectrolítico; algunas veces se requiere la perfusión endovenosa continua de líquidos porque la pérdida de estos puede ser hasta de 500 mL/kg/día, y la de sodio hasta de 45 mEq/kg/día; estos pacientes no requieren inicialmente suplementos de potasio pero se pueden beneficiar de ellos, y de la administración de indometacina, a partir de la cuarta a sexta semanas de vida.43 Esta terapia permite la estabilización clínica y lograr un crecimiento adecuado.
Entre los medicamentos utilizados están los diuréticos ahorradores de potasio como la espironolactona; sin embargo, esta droga puede aumentar la hipercalciuria y por tanto la nefrocalcinosis. La indometacina (1,5–2,5 mg/kg/día) es el medicamento más utilizado para tratar a estos pacientes porque hace disminuir la pérdida de sales y la alcalosis y mejora la capacidad para concentrar la orina; sin embargo, existe el riesgo de sufrir enterocolitis necrosante y disminución de la filtración glomerular, por lo cual no se la recomienda en prematuros, o debe iniciársela después de 4 a 6 semanas de vida con un control estricto de los signos de enterocolitis.
Síndrome de Bartter clásico (tipo 3)
El objetivo principal del tratamiento es corregir la hipokalemia y la alcalosis; siempre es necesaria la administración de cloruro de potasio, cuya cantidad se debe individualizar; en muchas ocasiones esto no es suficiente y se requiere usar también espironolactona o triamtireno;43 sin embargo, el efecto es solamente transitorio. Los medicamentos más benéficos para estos pacientes son los inhibidores de la síntesis de prostaglandinas tales como la indometacina (2–5 mg/kg/día); con ella se observan mejoría de la fuerza, disminución de la poliuria y la polidipsia, recuperación de la velocidad de crecimiento e incremento del nivel plasmático de potasio; su eficacia es a largo plazo y puede utilizársela por períodos prolongados.23 Ocasionalmente estos pacientes pueden presentar hipomagnesemia por lo que requieren suplementos de magnesio. Otras medicaciones utilizadas son el ácido acetilsalicílico, ibuprofeno y ketoprofeno.34 Con respecto a los inhibidores de la enzima convertidora de angiotensina, los estudios han mostrado resultados contradictorios por lo que no se utilizan en la actualidad; además existe el riesgo de hipotensión sintomática.
Síndrome de Bartter tipo 4
El tratamiento de esta variante neonatal es similar al anteriormente descrito; sin embargo, estos pacientes son especialmente vulnerables a la falla renal luego de la terapia con indometacina, por lo cual se debe recurrir a otras opciones terapéuticas como la espironolactona.51
Síndrome de Gitelman
El objetivo principal del tratamiento es corregir la hipomagnesemia; generalmente no es necesario administrar potasio ni inhibidores de las prostaglandinas. La presentación más utilizada es el cloruro de magnesio para así compensar las pérdidas de cloro. En raras ocasiones se requiere administrar sales de potasio o espironolactona para corregir la hipokalemia. Excepcionalmente es preciso administrar dosis altas de indometacina para evitar el retardo del crecimiento.52 En conclusión, los síndromes de Bartter y Gitelman son tubulopatías causadas por alteraciones en diferentes canales ubicados en el asa ascendente de Henle, que causan trastornos hidroelectrolíticos, los cuales se expresan en forma sistémica; en años recientes se han logrado grandes avances en la comprensión de su fisiopatología lo que abre la puerta a nuevas opciones de tratamiento; es importante tener en cuenta estos síndromes cuya baja frecuencia los hace difíciles de diagnosticar; el éxito del tratamiento depende de iniciarlo tempranamente.
PERSPECTIVAS
En las consultas de Nefrología Pediátrica del Hospital Pablo Tobón Uribe y de la Clínica Bolivariana, de Medellín, Colombia, se han diagnosticado estos síndromes en algunos pacientes pero, hasta la fecha no han sido clasificados. Los diagnósticos se basaron en el fenotipo y en los exámenes de laboratorio (pH y gases arteriales, ionograma, pruebas de función renal y ecografía renal). El objetivo es agruparlos en un servicio clínico para hacerles las caracterizaciones genética y fenotípica. De esta manera se logrará hacer diagnósticos tempranos y tratamientos adecuados y darles seguimiento y asesoría genética a los pacientes y sus familias.
REFERENCIAS BIBLIOGRÁFICAS
1. Flagg TP, Yoo D, Sciortino CM, Tate M, Romero MF, Welling PA. Molecular mechanism of a COOH–terminal gating determinant in the ROMK channel revealed by a Bartter's disease mutation. J Physiol 2002; 544: 351–362. [ Links ]
2. Rodríguez–Soriano J, Vallo A, Pérez de Nanclares G, Bilbao JR, Castaño L. A founder mutation in the CLCNKB gene causes Bartter syndrome type III in Spain. Pediatr Nephrol 2005; 20: 891–896. [ Links ]
3. Pennesi M, Marchetti F, Crovella S, Boaretto F, Travan L, Lazzerini M, et al. A new mutation in two siblings with cystinosis presenting with Bartter syndrome. Pediatr Nephrol 2005; 20: 217–219. [ Links ]
4. Chaudhuri A, Salvatierra O Jr, Alexander SR, Sarwal MM. Option of pre–emptive nephrectomy and renal transplantation for Bartter's syndrome. Pediatr Transplant 2006; 10: 266–270. [ Links ]
5. Vaisbich MH, Fujimura MD, Koch VH. Fujimura, Koch VH. Bartter syndrome: benefits and side effects of longterm treatment. Pediatr Nephrol 2004; 19: 858–863. [ Links ]
6. Konrad M, Weber S. Recent advances in molecular genetics of hereditary magnesium–losing disorders. J Am Soc Nephrol 2003; 14: 249–260. [ Links ]
7. Rudin A. Bartter's syndrome. A review of 28 patients followed for 10 years. Acta Med Scand 1988; 224: 165–171. [ Links ]
8. Shaer AJ. Inherited primary renal tubular hypokalemic alkalosis: a review of Gitelman and Bartter syndromes. Am J Med Sci 2001; 322: 316–332. [ Links ]
9. Zelikovic I. Hypokalaemic salt–losing tubulopathies: an evolving story. Nephrol Dial Transplant 2003; 18: 1696–1700. [ Links ]
10. Peters M, Ermert S, Jeck N, Derst C, Pechmann U, Weber S, et al. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int 2003; 64: 923–932. [ Links ]
11. Bailey MA, Cantone A, Yan Q, MacGregor GG, Leng Q, Amorim JB, et al. Maxi–K channels contribute to urinary potassium excretion in the ROMK–deficient mouse model of type II Bartter's syndrome and in adaptation to a high–K diet. Kidney Int 2006; 70: 51–59. [ Links ]
12. Cho JT, Guay–Woodford LM. Heterozygous mutations of the gene for Kir 1.1 (ROMK) in antenatal Bartter syndrome presenting with transient hyperkalemia, evolving to a benign course. J Korean Med Sci 2003; 18: 65–68. [ Links ]
13. Starremans PG, Kersten FF, Knoers NV, van den Heuvel LP, Bindels RJ. Mutations in the human Na–K–2Cl cotransporter (NKCC2) identified in Bartter syndrome type I consistently result in nonfunctional transporters. J Am Soc Nephrol 2003; 14: 1419–1426. [ Links ]
14. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na–K–2Cl cotransporter NKCC2. Nat Genet 1996; 13: 183–188. [ Links ]
15. Kitanaka S, Sato U, Maruyama K, Igarashi T. A compound heterozygous mutation in the BSND gene detected in Bartter syndrome type IV. Pediatr Nephrol 2006; 21: 190–193. [ Links ]
16. Adachi M, Asakura Y, Sato Y, Tajima T, Nakajima T, Yamamoto T, et al. Novel SLC12A1 (NKCC2) mutations in two families with Bartter syndrome type 1. Endocr J 2007; 54: 1003–1007. [ Links ]
17. Nüsing RM, Treude A, Weissenberger C, Jensen B, Bek M, Wagner C, et al. Dominant role of prostaglandin E2 EP4 receptor in furosemide–induced salt–losing tubulopathy: a model for hyperprostaglandin E syndrome/antenatal Bartter syndrome. J Am Soc Nephrol 2005; 16: 2354–2362. [ Links ]
18. Krishnan SN, Desai T, Ward DC, Haddad GG. Isolation and chromosomal localization of a human ATP–regulated potassium channel. Hum Genet 1995; 96: 155–160. [ Links ]
19. Kumar PS, Deenadayalan M, Janakiraman L, Vijayakumar M. Neonatal Bartter syndrome. Indian Pediatr 2006; 43: 735–737. [ Links ]
20. Pressler CA, Heinzinger J, Jeck N, Waldegger P, Pechmann U, Reinalter S, et al. Late–onset manifestation of antenatal Bartter syndrome as a result of residual function of the mutated renal Na+–K+–2Cl– co–transporter. J Am Soc Nephrol 2006; 17: 2136–2142. [ Links ]
21. Starremans PG, Van der Kemp AW, Knoers NV, Van den Heuvel LP, Bindels RJ. Functional implications of mutations in the human renal outer medullary potassium channel (ROMK2) identified in Bartter syndrome. Pflugers Arch 2002; 443: 466–472. [ Links ]
22. Crosara–Ayres C, Simoes e Silva A. Bartter's syndrome: five cases with different clinical expression. Jornal de Pediatria 2003; 79: 471–472. [ Links ]
23. Ammenti A, Montali S. 'Neonatal variant' of Bartter syndrome presenting with acidosis. Pediatr Nephrol 1996; 10: 79–80. [ Links ]
24. Fukuyama S, Hiramatsu M, Akagi M, Higa M, Ohta T. Novel mutations of the chloride channel Kb gene in two Japanese patients clinically diagnosed as Bartter syndrome with hypocalciuria. J Clin Endocrinol Metab 2004; 89: 5847–5850. [ Links ]
25. Nozu K, Fu XJ, Nakanishi K, Yoshikawa N, Kaito H, Kanda K, et al. Molecular analysis of patients with type III Bartter syndrome: picking up large heterozygous deletions with semiquantitative PCR. Pediatr Res 2007; 62: 364–369. [ Links ]
26. Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens 2003; 12: 527–532. [ Links ]
27. Duman O, Koyun M, Akman S, Güven AG, Haspolat S. Case of Bartter syndrome presenting with hypokalemic periodic paralysis. J Child Neurol 2006; 21: 255–256. [ Links ]
28. Bettinelli A, Borsa N, Bellantuono R, Syrén ML, Calabrese R, Edefonti A. Patients with biallelic mutations in the chloride channel gene CLCNKB: long–term management and outcome. Am J Kidney Dis 2006; 49: 91–98. [ Links ]
29. Briet M, Vargas–Poussou R, Lourdel S, Houillier P, Blanchard A. How Bartter's and Gitelman's syndromes, and Dent's disease have provided important insights into the function of three renal chloride channels: ClCKa/b and ClC–5. Nephron Physiol 2006; 103: 7–13. [ Links ]
30. Watanabe T, Tajima T. Renal cysts and nephrocalcinosis in a patient with Bartter syndrome type III. Pediatr Nephrol 2005; 20: 676–678. [ Links ]
31. Calo LA. Vascular tone control in humans: insights from studies in Bartter's/Gitelman's syndromes. Kidney Int 2006; 69: 963–966. [ Links ]
32. Tajima T, Nawate M, Takahashi Y, Mizoguchi Y, Sugihara S, Yoshimoto M, et al. Molecular analysis of the CLCNKB gene in Japanese patients with classic Bartter syndrome. Endocr J 2006; 53: 647–652. [ Links ]
33. Krämer BK, Bergler T, Stoelcker B, Waldegger S. Mechanisms of disease: the kidney–specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat Clin Pract Nephrol 2008; 4: 38–46. [ Links ]
34. Brennan TM, Landau D, Shalev H, Lamb F, Schutte BC, Walder RY, et al. Linkage of infantile Bartter syndrome with sensorineural deafness to chromosome 1p. Am J Hum Genet 1998; 62: 355–361. [ Links ]
35. Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM, Maier–Lutz I, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 2001; 29: 310–314. [ Links ]
36. Zaffanello M, Taranta A, Palma A, Bettinelli A, Marseglia GL, Emma F. Type IV Bartter syndrome: report of two new cases. Pediatr Nephrol 2006; 21: 766–770. [ Links ]
37. Schlingmann KP, Konrad M, Jeck N, Waldegger P, Reinalter SC, Holder M, et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 2004; 350: 1314–1319. [ Links ]
38. Miyamura N, Matsumoto K, Taguchi T, Tokunaga H, Nishikawa T, Nishida K, et al. Atypical Bartter syndrome with sensorineural deafness with G47R mutation of the beta–subunit for ClC–Ka and ClC–Kb chloride channels, barttin. J Clin Endocrinol Metab 2003; 88: 781–786. [ Links ]
39. Nozu K, Inagaki T, Fu XJ, Nozu Y, Kaito H, Kanda K, et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J Med Genet 2008; 45: 182–186. [ Links ]
40. Reinalter SC, Jeck N, Peters M, Seyberth HW. Pharmacotyping of hypokalaemic salt–losing tubular disorders. Acta Physiol Scand 2004; 181: 513–521. [ Links ]
41. Riveira–Munoz E, Chang Q, Bindels RJ, Devuyst O. Gitelman's syndrome: towards genotype–phenotype correlations? Pediatr Nephrol 2007; 22: 326–332. [ Links ]
42. Hayashi M. Gitelman's syndrome and hypomagnesemia. Intern Med 2004; 43: 351–352. [ Links ]
43. Unwin RJ, Capasso G. Bartter's and Gitelman's syndromes: their relationship to the actions of loop and thiazide diuretics. Curr Opin Pharmacol 2006; 6: 208–213. [ Links ]
44. Schlingmann KP, Konrad M, Seyberth HW. Genetics of hereditary disorders of magnesium homeostasis. Pediatr Nephrol 2004; 19: 13–25. [ Links ]
45. Pachulski RT, Lopez F, Shara R. Gitelman's not–so–benign syndrome. N Engl J Med 2005; 353: 850–851. [ Links ]
46. Bettinelli A, Silvana T. Hypokalemia and hypomagnesemia of hereditary renal tubular origin. Bartter and Gitelman syndromes. Acta Biomed Ateneo Parmense 2003; 74: 163–167. [ Links ]
47. Bettinelli A, Borsa N, Syrén ML, Mattiello C, Coviello D, Edefonti A, et al. Simultaneous mutations in the CLCNKB and SLC12A3 genes in two siblings with phenotypic heterogeneity in classic Bartter syndrome. Pediatr Res 2005; 58: 1269–1273. [ Links ]
48. Fernández R, Rodríguez JA, Barrera F, Weinberger J. Diagnóstico del síndrome de Bartter por estudio sistemático de la hipokalemia. Rev Chilena Pediatr 1982; 53: 229–233. [ Links ]
49. Proesmans W. Threading through the mizmaze of Bartter syndrome. Pediatr Nephrol 2006; 21: 896–902. [ Links ]
50. Colussi G, Bettinelli A, Tedeschi S, De Ferrari ME, Syrén ML, Borsa N, et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol 2007; 2: 454–460. [ Links ]
51. Ozlu F, Yapicioðlu H, Satar M, Narli N, Ozcan K, Buyukcelik M, et al. Barttin mutations in antenatal Bartter syndrome with sensorineural deafness. Pediatr Nephrol 2006; 21: 1056–1057. [ Links ]
52. Liaw LC, Banerjee K, Coulthard MG. Dose related growth response to indometacin in Gitelman syndrome. Arch Dis Child 1999; 81: 508–510. [ Links ]
Recibido: mayo 21 de 2008
Aceptado: septiembre 16 de 2008

